一 : 朱瑞新着-- 计算机辅助药物设计(Ⅰ)--基本方法原理
计算机辅助药物设计
------基本方法原理概要与实践详解
作者 朱瑞新
2011年1月
目录
序
前言
第一章 “计算机辅助药物设计”与MOE概貌
一、导言
二、“计算机辅助药物设计”概貌
三、MOE概貌
四、知识拓展
五、本章小结
六、复习思考题
第二章 虚拟组合库设计
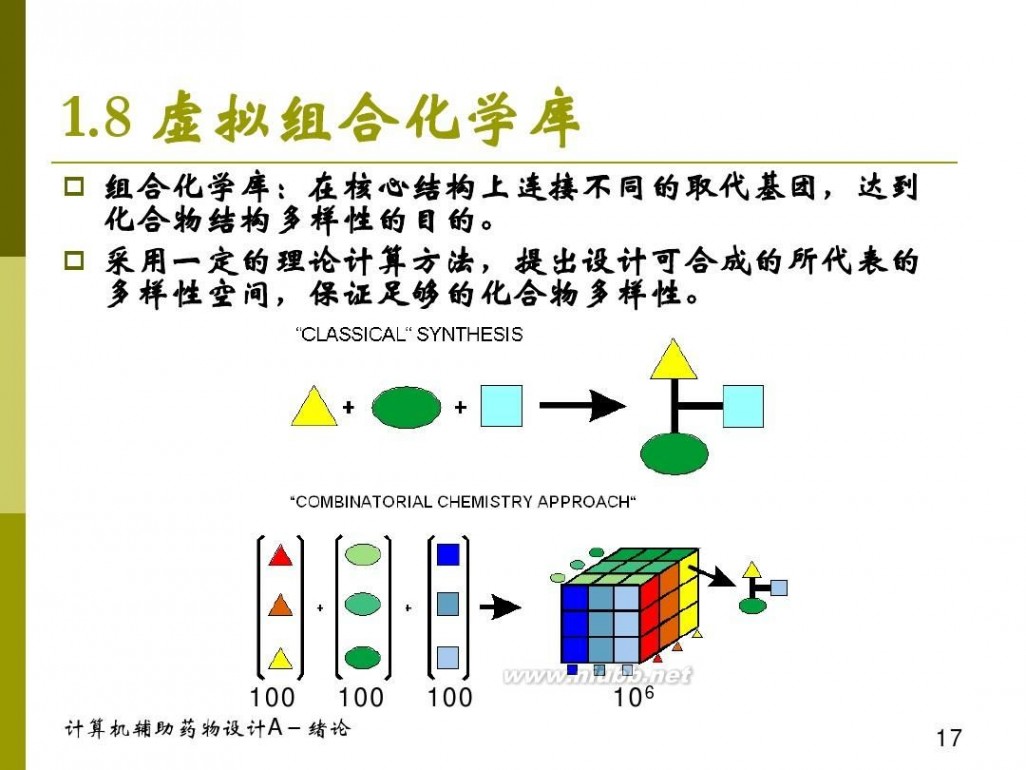
一、虚拟组合库设计的运用
二、组合化学与虚拟组合库设计
三、虚拟组合库设计的方法原理
四、虚拟组合库设计的操作要领
五、知识拓展
六、本章小结
七、复习思考题
第三章 生物大分子结构预测
一、生物大分子结构预测的运用
二、生物大分子结构测定与生物大分子结构预测
三、生物大分子结构预测的方法原理
四、生物大分子结构预测的操作要领
五、知识拓展
六、本章小结
七、复习思考题
第四章 定量构效关系
1
一、定量构效关系的运用
二、构效关系与定量构效关系
三、定量构效关系的方法原理
四、定量构效关系的操作要领
五、知识拓展
六、本章小结
七、复习思考题
第五章 药效团模型
一、药效团模型的运用
二、活性特征基团和药效团
三、药效团模型的方法原理
四、药效团模型的操作要领
五、知识拓展
六、本章小结
七、复习思考题
第六章 分子对接
一、分子对接的运用
二、分子(生化)水平的高通量筛选技术和分子对接
三、分子对接的方法原理
四、分子对接的操作要领
五、知识拓展
六、本章小结
七、复习思考题
第七章 全新药物设计
一、全新药物设计的运用
二、片段组学(基于片段的药物设计)和全新药物设计
三、全新药物设计的方法原理
四、全新药物设计的操作要领
五、知识拓展
2
六、本章小结
七、复习思考题
第八章 分子动力学、随机动力学和蒙特卡洛
一、分子动力学、随机动力学和蒙特卡洛的运用
二、体系全部性质测定和分子动力学/随机动力学/蒙特卡洛
三、分子动力学、随机动力学和蒙特卡洛的方法原理
四、分子动力学、随机动力学和蒙特卡洛的操作要领
五、知识拓展
六、本章小结
七、复习思考题
第九章 MOE中其它实用模块
一、受体-配体复合物指纹(PLIF)
二、大分子质子化(Protonate3D)
三、抗体模建(Antibody Modeler)
四、知识拓展
五、本章小结
六、复习思考题
参考文献
致谢
3
前言
本书系作者在近年来为同济大学本科生开设的《计算机辅助药物设计》课程所用讲义基础上,经扩展、补充和修改编写而成。
本书内容涵盖了“计算机辅助药物设计”七大研究方向:1、虚拟小分子生成、2、大分子结构预测、3、定量构效关系、4、药效团模型、5分子对接、6、全新药物设计和7、动态模拟(分子动力学/随机动力学/蒙特卡洛)。并且第一次系统地对这些技术的操作进行了讲解。其中,本书第一章是对“计算机辅助药物设计”和配合本书实例讲解的国际著名软件MOE的全貌进行了介绍;第二章到第八章依次对“计算机辅助药物设计”的七大研究方向进行了理论和实践操作的系统介绍;最后一章对MOE中三个特色模块进行了单独介绍。另外,本书每章还设有一个“知识拓展”,该部分主要是介绍一些更为前沿或热门的知识,希望能为读者的相关研究或深入学习起到抛砖引玉的效果。
“计算机辅助药物设计”是门非常综合且前沿的学科,对于本科生而言系统的理论学习将会严重打击其学习的积极性;同样如果只是科普性的介绍一些新概念或者介绍一些传奇故事,而不进行系统的实践操作,那么学生们的动手能力无法得到很好的训练,这点对于部分本科毕业即将要走上工作岗位的同学显得尤为重要。因此本书对于理论部分的介绍尽量做到简明扼要,而对于实践操作的讲解尽量做到详细,以期能够让读者一直保持着“强烈的掌握欲望”快速掌握“计算机辅助药物设计”核心技术。
本书是国内第一本专门为本科生撰写的《计算机辅助药物设计》教材,此外,本书也将是工业界相关人员和研究生的一本非常合适的参考书。
鉴于作者学识浅薄,再加上时间紧迫,书中定有多处不足,甚至错误之处,还请各位专家和读者指出。
朱瑞新
同济大学生命科学与技术学院
2011-01-05
第一章
本章内容目录
一、
二、
三、
四、
五、
六、
一、导言 导言 “计算机辅助药物设计”与MOE概貌 “计算机辅助药物设计”概貌 MOE概貌 知识拓展 本章小结 复习思考题
“数学就是一门把不同的东西归结为相同问题的艺术。”——Jules Henri Poincaré(法国数学物理学家,“最后一位数学通才”)
“用物理的火炬照亮化学的暗室。” ——Friedrich Wilhelm Ostwald (1909年Nobel化学奖得主,物理化学学科奠基人)
“计算化学家的时代即将到来。到时,若不是成千也有上百的化学家,为了化学中日益增多的许多细节问题将走向计算机,而不是去实验室。”——Robert Sanderson Mulliken (1966年获诺贝尔化学奖,分子轨道理论的创立者


)
时间?
图1-1、实验学科与相关计算学科发展的关系示意图
“计算”学科的诞生往往是晚于相应的实验学科(如图1-1所示),但也是相应实验学科发展迈向成熟的一个重要标志。回顾学科发展历史,各实验学科的深入发展都离不开数学和物理这两个“最基础”的学科辅助,同样,作为与人类健康息息相关的药物研发也离不开这两个学科的辅助。
上世纪中期诞生的计算机无疑是个划时代的成果。它的出现使得各个学科又一次发生了 1
全新的变化。比如,1953年著名的Fermi-Pasta-Ulam的计算机实验,研究了动力学体系非线性项的微扰是如何改变单一的周期振动行为的。结果出人意外:竟然恢复初始状态的时间远远比想象的Poincare回复时间短得多。这个计算机实验开创了“计算物理”这门新学科。然而这个实验产生的更为重要结果是:从此以后,人们明白除了实验、形式理论这两条能够创造、发现新的科学概念的途径之外,还存在第三条途径——模拟计算。有时候,一个演绎表达式不能让科学家立刻感悟到其中隐藏的科学概念,但是可以通过模拟计算发现它。
尽管如此,计算相关的学科在发展早期都或多或少的受到从事相关领域实验科学研究的工作者们怀疑或者忽视。直到1998年诺贝尔化学奖授予了Walter Kohn(提出密度泛函理论)和John A. Pople(提出波函数方法)两位专家后,计算化学/分子模拟等研究工作越来越受到认可和重视。
二、“计算机辅助药物设计”概貌
“计算机辅助药物设计”(computer aided drug design / computer assisted drug design)以计算机为载体,综合运用物理、化学和生物等学科的基本原理和(统计)数学知识,对药物研发的各个阶段进行辅助解释、预测和设计。
无论是按照Horst Friihbeis等人的说法(Angew. Chem. Int. Ed. Engl. 26(1987) 403-418):Cyrus Leuinthal于1966发表了第一篇关于“计算机辅助分子图形”研究论文(C. Levinthal, Sci. Am. 214 (1966) 42.)算起;还是按照:定量构效关系(Quantitative structure-activity relationship,QSAR)是“计算机辅助药物设计”中发展最早且成熟的方法,C. Hansch于1962年发表的Hansch方程(Nature, 1962, 194, 178)被认为是第一个可以实施的QSAR方法算起;计算机辅助“药物”设计的发展都已经有了半个世纪之久。但是作为一门独立且系统的学科,“计算机辅助药物设计”形成于上世纪八十年代,当时涌现出多篇至今看来都非常不错的综述(本书所带光盘附有其中几篇)。
自上世纪八十年代起,“计算机辅助药物设计”相关领域得到迅速发展。各种算法及软件日新月异,但是,所有的研究均可以归属于:1、虚拟小分子生成、2、大分子结构预测、
3、定量构效关系、4、药效团模型、5分子对接、6、全新药物设计和7、动态模拟等七大方向之一(如图1-2所示)。比如现在很多商业软件中提供的分子新陈代谢及毒性研究(ADMET)无非是定量构效关系研究的一个特例。本书自第二章开始,将依次介绍“计算机辅助药物设计”中的七大研究方向。
药物研究有两个研究对象:药物和受体。正是围着这两个研究对象,根据受体结构是否 2
已知和活性数据是否定量,使得“计算机辅助药物设计”研究经过长期的发展后,定格成这样的七大研究方向:在虚拟筛选时,需要产生大量的候选分子,这个需求导致产生了“虚拟小分子生成”这个研究领域;受实验测定限制,大量的受体结构信息需要利用计算机模拟的方法得到,这就催生了“大分子结构预测”这个研究领域;早期受体结构信息缺乏,但是围绕着同一受体或者疾病已经获得多个小分子的活性信息,根据该活性信息是否为定量信息,分别发展出定量的“定量构效关系”和半定量的“药效团模型”两个研究领域;随着近年来受体结构信息逐渐增多,诞生了“分子对接”这个在上世纪九十年代风靡一时的技术,与此同时,随着片段组学及前面几个“计算机辅助药物设计”技术的成熟发展及改进,催生了“全新药物设计”技术;“分子动力学/随机动力学/蒙特卡洛”是个非常特殊的技术,尽管该方法“不能”直接判断出某分子是否为药物候选分子,但是在前面六个技术中根据需要都将用到该方法,特别是在含有大分子结构的时候,是对整个分子体系进行优化使其到达某个“合理结构”状态不可或缺的方法。值得一提的是:早期的论文和专著中往往将“药效团模型”只归属于“基于配体”的方法,而事实上根据受体结构信息,计算受体活性位点处相应的表面性质,然后根据受体-配体之间的性质和空间结构互补特性,同样可以获得相应的“药效团模型”,也就是说“药效团模型”也可以是一种“基于受体”的方法,详见本书第五章。
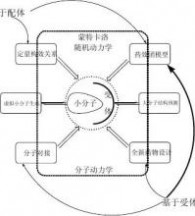
图1-2、“计算机辅助药物设计”七大研究方向
传统药物研发是个非常耗时、耗钱和劳动力密集型的工作,“计算机辅助药物设计”的引入对整个研发过程都有一定的“辅助”,有时候甚至起到推动药物研发或者决定药物研发成败的关键因素。感兴趣的读者可以阅读Tanaji T. Talele等人近期的综述(Current Topics in 3
Medicinal Chemistry, 2010, 10, 127-141),其中对1、Captopril (Capoten?, Bristol
Myers-Squibb),2、Dorzolamide (Trusopt?, Merck),3、Saquinavir (Invirase?, Hoffmann-La Roche),4、Zanamivir (Relenza?, Gilead Sciences),5、Oseltamivir (Tamiflu?, Gilead Sciences),
6、Aliskiren (Tekturna?, Novartis),7、Boceprevir (Schering-Plough),8、Nolatrexed
dihydrochloride (Thymitaq?, Agouron),9、TMI-005,10、LY-517717 (Lilly/Protherics),11、Rupintrivir (AG7088, Agouron),和12、NVP-AUY922 (Novartis)等12个利用“计算机辅助药物设计”方法发现或者优化并得到美国食品安全局批准的药物进行了介绍。读者还可以进一步阅读德国著名药物学家Hugo Kubinyi于2006年撰写的专著(Computer Applications in
Pharmaceutical Research and Development, Ekins, S. Ed. Wiley-Interscience, NY,2006年),从而获得对“计算机辅助药物设计”应用的更多自信。
这里特别要提醒读者的是,药物研究是个非常复杂,并且风险很高的事情,本书仅仅是为了方便读者的理解,才将“计算机辅助药物设计”研究分解成如上图1-2所示的七大研究方向。在实际研究中,往往需要综合运用上述研究方向的多个或全部核心技术。
例如在对药物进行高通量虚拟筛选,往往需要进行如下步骤过滤(摘自www.kubinyi.de):
1、 是否有书写错误等(Garbage filter);
2、 是否有类药性(Druglike / Non-druglike);
3、 是否达到一定的生物利用度(Bioavailability):定量构效关系和药效团模型;
:定量构效关系和药效团模型; 4、 是否具有细胞毒性(Cytotoxicity)
:定量构效关系、药5、 是否抑制hERG钾离子通道(hERG channel inhibition)
效团模型和分子对接;
6、 是否与下列常见抗靶标有作用(Antitargets):
(1)?1a (orthostatic hypotension);
(2)D2 (extrapyramidal syndrome);
(3)5-HT2c (obesity);
(4)musc. M1 (hallucinations, memory);
定量构效关系、药效团模型、同源模建和分子对接;
7、 是否对P450代谢酶有抑制作用(CYP inhibition (3A4, 2C9, 2D6)):定量构
效关系、药效团模型和分子对接。
在这样一个通用的药物虚拟高通量筛选过程中,就需要直接的综合运用大分子结构预 4
测、定量构效关系、药效团模型和分子对接四种方法;并且需要间接的运用同源模建、分子库设计和动力学模拟三种方法。
本节最后简要地对“计算机辅助药物设计”所用到的方法基础进行概括。更为详细的参考请阅读中国科学院上海有机化学研究所陈敏伯研究员近期撰写的《计算化学:从理论化学到分子模拟》(科学出版社,2009年版)。需要强调的是:本书无意对“计算机辅助药物设计”方法中涉及到的纯粹数学方法作深入介绍,作者认为对这些内容的过多讲解往往分散读者的注意力,最终导致读者不能很好的从整体上了解和掌握“计算机辅助药物设计”相关知识。
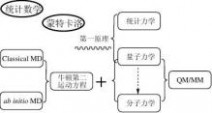
图1-3、“计算机辅助药物设计”的方法基础
如上图(图1-3)所示,同其它自然科学一样,“计算机辅助药物设计”的基础理论也是建立在“数学”和“物理”这两个最基础的学科之上。
1、 数学
除了常规的数学知识外,“计算机辅助药物设计”的多个核心技术对统计数学和蒙特卡洛十分偏爱,尤其是定量构效关系研究中,各种统计数学方法的引入让该技术从诞生至今仍受到广泛的重视(详见本书第四章),而在构象优化等问题上,蒙特卡洛算法一直是讨论的热点。
2、 物理
“事实上,量子力学规律不仅支配着微观世界,而且也支配着宏观世界,可以说全部物理学都是量子物理学的。已被长期实践证明的描述宏观现象的经典力学规律,实质上不过是量子力学规律的一个近似。”
简单来讲,“量子力学”是研究微观粒子各种属性的物理方法;“统计力学”则是将粒子们的微观属性同它的宏观表现关联起来的物理方法;而一切基于“量子力学”和“统计力学”演绎而得到的方法就是所谓的“第一原理”或者“从头算原理”方法。量子力学发展至今,尚未得出矛盾的结论,这样一来,便有了“从第一原理出发就可以解释一切现象”的观点。 5
量子力学的发展,根据是否直接计算电子的波函数而形成了两大类研究方法:(1)、波函数和(2)、密度泛函。1998年诺贝尔化学奖就是分别授予了这两类方法的代表人物John A. Pople和Walter Kohn。
可是,令人沮丧的是,即使选用计算量较低的密度泛函方法,对于生物大分子来讲仍然是不大现实的。而前面提到,作为药物受体的典型代表之一的生物大分子,在药物设计研究中显然是无法避免的。
于是类似于AM1、PM3和MNDO这些半经验的“从头算”方法不断地出现并一度成为该领域的通用方法。概括来讲,这些半经验方法都属于波函数派系,利用一定的实验数据信息拟合成参数简化原来“第一原理”相关方程中的部分项,比如利用高斯函数替代“原子实”的时候。
与此同时,LS-DFT(Linear-Scaling Density Functional Theory, 线性密度泛函法)等基于简化“第一原理”中密度泛函计算模型的方法也得到发展。
即便是使用半经验量化和线性密度泛函法,对于生物大分子的研究仍然不是很现实,在这样的基础上,大家不得转而使用对量子力学完全参数化的分子力学方法,由于分子力学方法中最关键的是各种简化的分子力场参数,因此这种方法经常也叫分子力场法。该方法将分子的能量可以近似看作构成分子的各个原子的空间坐标的函数,描述这种分子能量和分子结构之间关系的就是分子力场函数, 而分子力场函数来自实验结果的经验公式。
分子力场方法的使用使得大分子体系的研究得到了迅速发展,分子力场方法的计算量相对量子力学方法而言要小数十倍,而且在适当的精度内,分子力场方法得出的结果与量子力学计算结果相差无几。但是要注意的是:分子力场方法在研究分子中含有键的生成或断裂时是不适用的。这是分子力学方法的先天不足之处。
药物设计中经常会要研究大分子和小分子相互作用的复合体系。为了兼顾小分子与大分子相互作用界面的精度和计算量大小,将量子力学(QM)和分子力学(MM)杂合在一起用于体系的研究,即在相互作用界面采用精度高的量子力学进行研究,而在远离相互作用界面的地方(主要是大分子的部分残基)使用计算量小的分子力学进行研究。这样就诞生了QM/MM方法。
分子动力学、随机动力学和蒙特卡洛方法都可以用于计算体系的热力学量和其他宏观性质,分子动力学是以各种力学(包括经典力学和量子力学)和61阅读络类方法再试(当然一般在实际的药物设计中,并不建议做到这一步)。
比如,此例中,我们首选进行剔除Outlier操作。
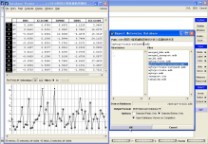
图4-15、剔除Outlier
具体操作:在训练集数据库中,首先DBV|Display,并勾上Plot,在Plot Fields中勾上$Z-SCORE,然后选择Z-SCORE中小于2的样本(剔除Outlier),单独保存为“mytrps-train-refined1.mdb”数据库,如图4-15所示。
然后在新产生的训练集的基础上重复步骤(4)。

图4-16、第二次训练集自身交叉验证的结果
80
二 : 关于计算机辅助药物设计的几点思考(转载)
引自科学网:摘要:
本文讨论了计算机辅助药物设计中的若干问题,仅为一家之言,供参考。
1.LBDD和SBDD方法的对应
2.LBDD和SBDD方法的整合
3.药物设计方法的层次
4.关于虚拟筛选
5.关于药物设计软件
6.CADD研究的困难
关键词:计算机辅助药物设计;讨论;CADD;LBDD;SBDD;
计算机辅助药物设计(computer-aided drugdesign,CADD)大大加快了药物发现的速度。广义的CADD泛指在信息技术在药物分子设计与开发过程中的所有应用,包含信息分析技术、数据处理过程等;而通常意义上的CADD仅指基于分子模拟(计算化学)的分子设计技术,又可分为基于受体结构的药物设计(receptor-based orstructure-based drug design, SBDD)和基于配体的药物设计(ligand-based drugdesign, LBDD)。SBDD从受体的结构和性质出发寻找可以与其特异结合的配体分子,所以也称为直接药物设计,包括基于受体结构的分子对接、活性位点分析、从头药物设计(生长或连接算法)等具体方法;LBDD根据已知活性的先导分子,构建结构-活性关系或药效基团模型,称为间接药物设计,包括定量构效关系(QSAR)、药效基团模型、受体映射、基于分子形状的叠合等具体的方法。在实际工作中,根据已知信息的多少可以选择相应的研究方法,当然已知信息越多越准确的时候,结果就越可靠。
本文结合作者从事相关工作的实践,谈一下对CADD的几点思考。
当然关于CADD本身的“有用”与“无用”的争辩,这里也就免了。(假舆马者,非利足也,而致千里;假舟楫者,非能水也,而绝江河。君子生非异也,善假於物也。(《荀子·劝学》))
1.LBDD和SBDD方法的对应
虽然SBDD和LBDD两种方法有完全不同的出发点,但是二者在方法学或设计思想上存在细微而有趣的对应关系。任伟同学将我在《药物设计概论》课程中的这个观点整理成了一篇论文——《基于配体和受体的药物设计方法的对应性》,发表在《生命科学仪器》(2009,7:19)上。在这里将这一对应简单整理如下表,供感兴趣的朋友参考:
Table1 Corresponding of receptor andligand based drug design methods
表1 基于受体和配体药物设计方法的对应
SBDDexample | LBDDexample | ||
Gridbased Step sized orrandom placed | Atomicprobe | Grid | CoMFA,COMSIA, SOMFA, MFA |
Fragment probe | MCSS,MFS | TopomaCoMFA | |
Docking | Traditional docking DOCK,FlexX, GOLD,AutoDock,FRED, SurflexDock,Glide | Reversed docking RT-Dock | |
FeatureBased | Pharmacophore based on protein Or Feature based protein sitesimilarity/classfication | Pharmacophore DISCO,Catalyst | |
Fragment based | Fragment searching Ludi,MFS, MCSS | Legend,HQSAR, TopmaCoMFA, Scafflod hopping, Fragment basedsimilarity, | |
Alignment or superimpose | Structure or sequence based proteinalignment | Corestructure, pharmacophore or field based alignment | |
Knowledge based | Target like Active site like Protein diversity universe | Drug like Lead Like Diversity | |
Similarity ordrug-target network | Protein similaritylandscape | SAR-MAP and Chemical spaceanalysis | |
Hypothetical model | Initial design | Hypothetical receptor model HASL | |
下面的思维导图也供参考:

药物设计中SBDD和LBDD的对应甚至可以体现在整体水平上——蛋白质设计与改造vs. 配基设计。药物设计是根据蛋白质的结构设计小分子配体,蛋白质设计是设计或改造基因序列,使表达后的蛋白具有特定的功能(如结合或催化底物),二者都是基于分子之间的相互作用。蛋白质改性、设计是一个十分重要并充满挑战的领域,并有可能藉此推动生命起源研究的发展。在蛋白设计发展到一定阶段说不定会出现潜在活性的蛋白库(虚拟的或者实体商品库)。到那时,将会出现与现在配体库的分析相对应的蛋白质的定量结构功能关系、蛋白商品库的多样性分析、生物相关性、稳定性研究等等研究方向。
可见,SBDD、LBDD这两种出发点完全不同的药物设计方法确实存在着微妙的对应关系。认识到这种对应关系不仅为药物设计方法的发展研究提供了素材,而且更为重要的是为今后药物设计方法的创新提供了重要的提示。例如,可以根据某种SDBB方法设计对应的LBDD方法,反之亦然。这为今后的药物设计方法的开发指出了一个捷径。
2.LBDD和SBDD方法的整合
SBDD以锁匙模型为基础,寻找可以与靶标结合的配体分子;而SBDD以相似性原理为基础,寻找与已知抑制剂具有潜在相同作用方式的配体分子。SBDD结果准确,可直接预测结合能,缺点是需要受体结构,速度慢;LBDD速度快,无需蛋白结构,但是有时由于缺乏具体结合方式的指导,结果外推时常常误差很大。这两种方法、理论是独立发展,各有其适用条件和利弊的。同时,这两类方法也是紧密联系的,将两种方法结合使用,特别是基于分子对接的三维定量构效关系研究(CoMFA based onDocking)常见于报道。以下是部分相关的文献:
lAn Integrated Approach toLigand- and Structure-Based Drug Design: Development andApplication to a Series of Serine Protease Inhibitors. J. Chem. Inf. Model.2008, 48, 1211–1226
lCombining Structure-Based DrugDesign and Pharmacophores. Journal of MolecularGraphics and Modelling 23 (2005) 439–446
lPseudoreceptor models in drugdesign: bridging ligand- and receptor-based virtual screening.NRDD, 2008,7:667
lCombined Target-Based andLigand-Based Drug Design Approach as a Tool To Define a Novel3D-Pharmacophore Model of Human A3 Adenosine Receptor Antagonists:Pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine Derivatives as a KeyStudyJ. Med. Chem. 2005, 48, 152-162
lComparison of Ligand-Based andReceptor-Based Virtual Screening of HIV Entry Inhibitors for theCXCR4 and CCR5 Receptors Using 3D Ligand Shape Matching andLigand-Receptor Docking. J. Chem. Inf. Model., 48 (3), 509 -533,2008.
lA combined ligand-based andtarget-based drug design approach for G-protein coupled receptors:application to salvinorin A, a selective kappa opioid receptoragonist. J Comput Aided Mol Des (2006) 20:471–493
lCombining ligand-based andstructure-based drug design in the virtual screening arena. EODDJanuary 2007, Vol. 2, No. 1, Pages 37-49
LBDD和SBDD方法整合的趋势也体现在数据库的设计上,不仅大分子数据库加入了抑制剂等配基的信息,将小分子数据库中加入活性数据(PubChem,ChemBL,DrugBank)上,而且在系统生物学的高度上,将大小分子信息整合为一个生物代谢调控的网络,并用于药物设计和生物基础研究。
此外,不同的LBDD或SBDD方法之间还可以相互整合,如多拷贝子结构搜寻(MCSS,in Insight II orDiscovery Studio)、多片段搜寻(MFS,inMOE)是基于片段的药物设计方法和基于格点的药物设计方法的整合。基于药效团的QSAR是LBDD中内部结合使用的例子。现在将两种方法结合使用的趋势越来越受到重视。
下图仅供参考:
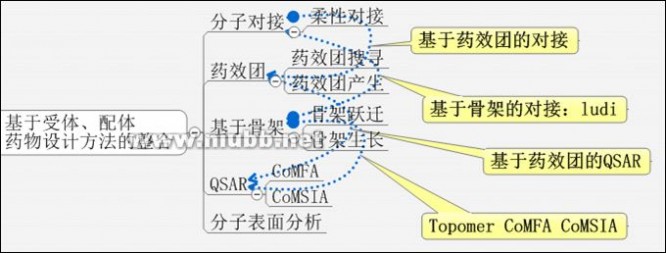
3.药物设计方法的层次
Grid是一个很有特色的方法,DDT上有一篇论文对此进行了讨论,S. Cross, G. Cruciani,Drug Discov Today 2010, 15(1-2), 23-32。我们都知道ComFA是用探针对小分子进行格点扫描。Grid软件是用格点方法扫描大分子。不过,用于蛋白质活性口袋的表征时,GRID软件不仅使用原子探针(如碳正离子),还使用分子片段探针(羟基、氨基、羧基、苯环等),甚至允许使用整个分子作为探针(类似于分子对接)。(多原子探针的方法还应用在Accelrys公司的Ludi、MCSS、CGC公司的MOE中MultiFragmentSearch(MFS)等模块上,但是它们都允许这些小分子片段运动/优化其位置。)
可见,药物设计分析方法存在着层次差异:
l电子:量化计算得到的静电势、电子云密度,轨道信息
l亚原子:Grid探针的点阵扫描
l原子:原子生长法,基于原子类型的LogP、MR计算;
l片段:片段连接法、HQSAR、骨架迁越、TopomaCoMFA、Hansch、FreeWilson
l分子:分子对接、分子描述符
相应地,我们都知道分子描述符可以分为:一维、二维、三维甚至多维;大分子的研究水平也可以分为一级序列(AA)、motif、二级结构和Fold、三级结构、结构域等水平。
//本来想花点时间,基于SBDD、LBDD的对应、整合以及药物设计层次的分类,整理出计算机辅助药物设计中QSAR、SBDD等方法的理论策略和技术发展的路线图,给出各种药物设计方法之间的联系(横向)及其发展脉络(纵向)。可是实在事情太多,而且能力有限,只好留爪在此,以后再说。
4.关于虚拟筛选
在药物设计中,大家常常会用到”虚拟筛选”这一名词,通常是指分子对接和基于药效基团的数据库搜寻。由于PDB数据库中蛋白质结构数量的快速增加,以及分子对接方法的进展,分子对接得到了全面的发展和广泛的应用,几乎成了虚拟筛选(VS)的代名词。其实,所有的CADD工作都可以看作虚拟筛选,即从众多的化合物(也包括虚拟的化合物)中得到活性的化合物。无论是分子对接、QSAR、药效基团、分子相似性筛选、三维结构叠合、分子骨架拆分等等都是虚拟筛选的途径。VS是目的,不是方法,CADD =VS。所有的SBDD都是基于互补的筛选,而所有的LBDD方法是基于相似性的筛选。各种筛选方法有不同的条件限制和速度,应采用先粗筛、再精筛的策略,在有限的时间内完成先导化合物的发现。
5.关于药物设计软件
药物设计中最重要的是人的经验,特别是药物化学、药理学基础知识的经验,而不全是软件的优劣。相同的软件在不同的学者手中发挥的作用是不同的。CADD离不开软件,一个软件就是一种思维方式、问题求解思路的体现。从这个意义上讲,尽管各种对接软件采用不同的搜索算法、结合能力评价方式,但是所有的分子对接的软件都具有相同的思想,即配体和蛋白质之间的互补。可能一个新的对接软件采用了更高效的算法、更准确的打分,解决的还是同一个问题。虽然筛选策略的创新需要技术方法的支持,但是,研究策略的创新显然比纯粹方法上的改进更有意义。
软件的学习要熟悉该软件的理论方法和应用条件,特别是软件的新思想,而不是软件的用法。随着时间的推移,软件的界面、选项、平台都有可能发生变化,仅仅是学习软件的使用方法意义不大。每个软件的力场和理论虽然有所差别,可是基本理论还是一样的,大同小异而已,只有学会软件的基本理论才能以不变应万变。因此,要注意软件背后的原理,熟悉软件的适用条件和体系。
药物设计软件正两个方向发展,一个是综合平台越来越多,所以专业综合软件的价格越来越低,从这个方面说,药物设计软件已经不是从事药物理论研究的瓶颈,几乎每一个研究所,每一个公司都可以支撑一个CADD项目组。另外一方面,单功能的药物设计软件的力量也在加强,有些甚至强大到和综合软件相抗衡的地步,如MolecularDiscovery的GRID历经多年依然年轻活力,而且衍生出一系列的应用(Volsurf等);此外,SARNavigator,Recore,Gold等软件,都是依靠其独特的算法、思路,专利的力量维持自己的发展,占据CADD软件市场一角。
药物设计软件中有免费和商业两种,对于制药公司,由于追求速度和效率,往往选择昂贵的商业软件,而对于科研用户,往往在二者之间徘徊,由于囊中羞涩,即向往于免费软件的自由,同时又觊觎商业软件操作的简便;虽然自由软件(特别是科研部门开发的软件)存在界面和操作上各种不足,但是其在应用领域的创新上,甚至是方法的创新上一直走在前列。以下是几个讨论文章:
lDrug Discov Today. 2005 Feb 1;10(3):219-22. Open-source software:not quite endsville.
lDrug Discov Today.2005 Feb 1;10(3):213-7. The case for open-source software in drugdiscovery.
lDrug Discov Today.2006 Feb;11(3-4):127-32. Optimizing the use of open-source softwareapplications in drug discovery.
lNat Rev DrugDiscov. 2006 Sep;5(9):723-9. Epub 2006 Aug 18. Can open-sourceR&D reinvigorate drug research?
关于生物信息学和化学信息学软件开源的讨论参看我前面的博文。
6.CADD研究的困难
10年前,从事CADD研究的困难在于软硬件,昂贵的SGI机器、昂贵的sybyl、InsightII很少研究组能够买的起;5年前,主流药物设计软件都移植到了Linux平台,尽管用户体验还不是很好,毕竟已经开始平民化了,这个时候CADD工作者的困难变成了如何将理论同实验结合,真正的“辅助药物设计”;现在,从事CADD研究的困难则更多的在于缺乏新的药物设计思想,国内外众多的研究组都盯着为数不多的已经验证或者富有潜力的靶标,要想在资金和积累都十分有限的条件下脱颖而出是很困难的。比如HIV-1蛋白酶,已知的抑制剂数以千计,复合物的晶体结构估计也快400个了(09年7月份查询的结果是357个),采用传统的方法,要想做出突破真的是不容易。

几个策略方向:
l药物设计最重要的是“靶标的选择”,靶标好,多花点钱,一定可以成功,靶标差,花了钱也会失败。现在的趋势是结合系统生物学、比较基因组学和网络药理学进行药物设计。
l多靶标药物设计(单药多靶、多药协同);
l数据挖掘(老药新用,DDT最近有一期专门讨论这个话题);
l基于片段的药物设计和骨架跃迁;
l高内涵筛选;
最后,以两幅忘了出处的图结束本文,以飨读者。


本文只为一家之言,由于知识、表达能力、时间有限,不足及错误之处难免,请原谅并不吝赐教。
三 : 朱瑞新着-- 计算机辅助药物设计(Ⅰ)--基本方法原理概要与实践详解
计算机辅助药物设计
------基本方法原理概要与实践详解
作者 朱瑞新
2011年1月
计算机辅助药物设计 朱瑞新着-- 计算机辅助药物设计(Ⅰ)--基本方法原理概要与实践详解
目录
序
前言
第一章 “计算机辅助药物设计”与MOE概貌
一、导言
二、“计算机辅助药物设计”概貌
三、MOE概貌
四、知识拓展
五、本章小结
六、复习思考题
第二章 虚拟组合库设计
一、虚拟组合库设计的运用
二、组合化学与虚拟组合库设计
三、虚拟组合库设计的方法原理
四、虚拟组合库设计的操作要领
五、知识拓展
六、本章小结
七、复习思考题
第三章 生物大分子结构预测
一、生物大分子结构预测的运用
二、生物大分子结构测定与生物大分子结构预测
三、生物大分子结构预测的方法原理
四、生物大分子结构预测的操作要领
五、知识拓展
六、本章小结
七、复习思考题
第四章 定量构效关系
1
计算机辅助药物设计 朱瑞新着-- 计算机辅助药物设计(Ⅰ)--基本方法原理概要与实践详解
一、定量构效关系的运用
二、构效关系与定量构效关系
三、定量构效关系的方法原理
四、定量构效关系的操作要领
五、知识拓展
六、本章小结
七、复习思考题
第五章 药效团模型
一、药效团模型的运用
二、活性特征基团和药效团
三、药效团模型的方法原理
四、药效团模型的操作要领
五、知识拓展
六、本章小结
七、复习思考题
第六章 分子对接
一、分子对接的运用
二、分子(生化)水平的高通量筛选技术和分子对接
三、分子对接的方法原理
四、分子对接的操作要领
五、知识拓展
六、本章小结
七、复习思考题
第七章 全新药物设计
一、全新药物设计的运用
二、片段组学(基于片段的药物设计)和全新药物设计
三、全新药物设计的方法原理
四、全新药物设计的操作要领
五、知识拓展
2
计算机辅助药物设计 朱瑞新着-- 计算机辅助药物设计(Ⅰ)--基本方法原理概要与实践详解
六、本章小结
七、复习思考题
第八章 分子动力学、随机动力学和蒙特卡洛
一、分子动力学、随机动力学和蒙特卡洛的运用
二、体系全部性质测定和分子动力学/随机动力学/蒙特卡洛
三、分子动力学、随机动力学和蒙特卡洛的方法原理
四、分子动力学、随机动力学和蒙特卡洛的操作要领
五、知识拓展
六、本章小结
七、复习思考题
第九章 MOE中其它实用模块
一、受体-配体复合物指纹(PLIF)
二、大分子质子化(Protonate3D)
三、抗体模建(Antibody Modeler)
四、知识拓展
五、本章小结
六、复习思考题
参考文献
致谢
3
计算机辅助药物设计 朱瑞新着-- 计算机辅助药物设计(Ⅰ)--基本方法原理概要与实践详解
前言
本书系作者在近年来为同济大学本科生开设的《计算机辅助药物设计》课程所用讲义基础上,经扩展、补充和修改编写而成。(www.61k.com)
本书内容涵盖了“计算机辅助药物设计”七大研究方向:1、虚拟小分子生成、2、大分子结构预测、3、定量构效关系、4、药效团模型、5分子对接、6、全新药物设计和7、动态模拟(分子动力学/随机动力学/蒙特卡洛)。并且第一次系统地对这些技术的操作进行了讲解。其中,本书第一章是对“计算机辅助药物设计”和配合本书实例讲解的国际著名软件MOE的全貌进行了介绍;第二章到第八章依次对“计算机辅助药物设计”的七大研究方向进行了理论和实践操作的系统介绍;最后一章对MOE中三个特色模块进行了单独介绍。另外,本书每章还设有一个“知识拓展”,该部分主要是介绍一些更为前沿或热门的知识,希望能为读者的相关研究或深入学习起到抛砖引玉的效果。
“计算机辅助药物设计”是门非常综合且前沿的学科,对于本科生而言系统的理论学习将会严重打击其学习的积极性;同样如果只是科普性的介绍一些新概念或者介绍一些传奇故事,而不进行系统的实践操作,那么学生们的动手能力无法得到很好的训练,这点对于部分本科毕业即将要走上工作岗位的同学显得尤为重要。因此本书对于理论部分的介绍尽量做到简明扼要,而对于实践操作的讲解尽量做到详细,以期能够让读者一直保持着“强烈的掌握欲望”快速掌握“计算机辅助药物设计”核心技术。
本书是国内第一本专门为本科生撰写的《计算机辅助药物设计》教材,此外,本书也将是工业界相关人员和研究生的一本非常合适的参考书。
鉴于作者学识浅薄,再加上时间紧迫,书中定有多处不足,甚至错误之处,还请各位专家和读者指出。
朱瑞新
同济大学生命科学与技术学院
2011-01-05
计算机辅助药物设计 朱瑞新着-- 计算机辅助药物设计(Ⅰ)--基本方法原理概要与实践详解
第一章
本章内容目录
一、
二、
三、
四、
五、
六、
一、导言 导言 “计算机辅助药物设计”与MOE概貌 “计算机辅助药物设计”概貌 MOE概貌 知识拓展 本章小结 复习思考题
“数学就是一门把不同的东西归结为相同问题的艺术。(www.61k.com)”——Jules Henri Poincaré(法国数学物理学家,“最后一位数学通才”)
“用物理的火炬照亮化学的暗室。” ——Friedrich Wilhelm Ostwald (1909年Nobel化学奖得主,物理化学学科奠基人)
“计算化学家的时代即将到来。到时,若不是成千也有上百的化学家,为了化学中日益增多的许多细节问题将走向计算机,而不是去实验室。”——Robert Sanderson Mulliken (1966年获诺贝尔化学奖,分子轨道理论的创立者


)
时间
图1-1、实验学科与相关计算学科发展的关系示意图
“计算”学科的诞生往往是晚于相应的实验学科(如图1-1所示),但也是相应实验学科发展迈向成熟的一个重要标志。回顾学科发展历史,各实验学科的深入发展都离不开数学和物理这两个“最基础”的学科辅助,同样,作为与人类健康息息相关的药物研发也离不开这两个学科的辅助。
上世纪中期诞生的计算机无疑是个划时代的成果。它的出现使得各个学科又一次发生了 1
计算机辅助药物设计 朱瑞新着-- 计算机辅助药物设计(Ⅰ)--基本方法原理概要与实践详解
全新的变化。[www.61k.com]比如,1953年著名的Fermi-Pasta-Ulam的计算机实验,研究了动力学体系非线性项的微扰是如何改变单一的周期振动行为的。结果出人意外:竟然恢复初始状态的时间远远比想象的Poincare回复时间短得多。这个计算机实验开创了“计算物理”这门新学科。然而这个实验产生的更为重要结果是:从此以后,人们明白除了实验、形式理论这两条能够创造、发现新的科学概念的途径之外,还存在第三条途径——模拟计算。有时候,一个演绎表达式不能让科学家立刻感悟到其中隐藏的科学概念,但是可以通过模拟计算发现它。
尽管如此,计算相关的学科在发展早期都或多或少的受到从事相关领域实验科学研究的工作者们怀疑或者忽视。直到1998年诺贝尔化学奖授予了Walter Kohn(提出密度泛函理论)和John A. Pople(提出波函数方法)两位专家后,计算化学/分子模拟等研究工作越来越受到认可和重视。
二、“计算机辅助药物设计”概貌
“计算机辅助药物设计”(computer aided drug design / computer assisted drug design)以计算机为载体,综合运用物理、化学和生物等学科的基本原理和(统计)数学知识,对药物研发的各个阶段进行辅助解释、预测和设计。
无论是按照Horst Friihbeis等人的说法(Angew. Chem. Int. Ed. Engl. 26(1987) 403-418):Cyrus Leuinthal于1966发表了第一篇关于“计算机辅助分子图形”研究论文(C. Levinthal, Sci. Am. 214 (1966) 42.)算起;还是按照:定量构效关系(Quantitative structure-activity relationship,QSAR)是“计算机辅助药物设计”中发展最早且成熟的方法,C. Hansch于1962年发表的Hansch方程(Nature, 1962, 194, 178)被认为是第一个可以实施的QSAR方法算起;计算机辅助“药物”设计的发展都已经有了半个世纪之久。但是作为一门独立且系统的学科,“计算机辅助药物设计”形成于上世纪八十年代,当时涌现出多篇至今看来都非常不错的综述(本书所带光盘附有其中几篇)。
自上世纪八十年代起,“计算机辅助药物设计”相关领域得到迅速发展。各种算法及软件日新月异,但是,所有的研究均可以归属于:1、虚拟小分子生成、2、大分子结构预测、
3、定量构效关系、4、药效团模型、5分子对接、6、全新药物设计和7、动态模拟等七大方向之一(如图1-2所示)。比如现在很多商业软件中提供的分子新陈代谢及毒性研究(ADMET)无非是定量构效关系研究的一个特例。本书自第二章开始,将依次介绍“计算机辅助药物设计”中的七大研究方向。
药物研究有两个研究对象:药物和受体。正是围着这两个研究对象,根据受体结构是否 2
计算机辅助药物设计 朱瑞新着-- 计算机辅助药物设计(Ⅰ)--基本方法原理概要与实践详解
已知和活性数据是否定量,使得“计算机辅助药物设计”研究经过长期的发展后,定格成这样的七大研究方向:在虚拟筛选时,需要产生大量的候选分子,这个需求导致产生了“虚拟小分子生成”这个研究领域;受实验测定限制,大量的受体结构信息需要利用计算机模拟的方法得到,这就催生了“大分子结构预测”这个研究领域;早期受体结构信息缺乏,但是围绕着同一受体或者疾病已经获得多个小分子的活性信息,根据该活性信息是否为定量信息,分别发展出定量的“定量构效关系”和半定量的“药效团模型”两个研究领域;随着近年来受体结构信息逐渐增多,诞生了“分子对接”这个在上世纪九十年代风靡一时的技术,与此同时,随着片段组学及前面几个“计算机辅助药物设计”技术的成熟发展及改进,催生了“全新药物设计”技术;“分子动力学/随机动力学/蒙特卡洛”是个非常特殊的技术,尽管该方法“不能”直接判断出某分子是否为药物候选分子,但是在前面六个技术中根据需要都将用到该方法,特别是在含有大分子结构的时候,是对整个分子体系进行优化使其到达某个“合理结构”状态不可或缺的方法。[www.61k.com]值得一提的是:早期的论文和专著中往往将“药效团模型”只归属于“基于配体”的方法,而事实上根据受体结构信息,计算受体活性位点处相应的表面性质,然后根据受体-配体之间的性质和空间结构互补特性,同样可以获得相应的“药效团模型”,也就是说“药效团模型”也可以是一种“基于受体”的方法,详见本书第五章。

图1-2、“计算机辅助药物设计”七大研究方向
传统药物研发是个非常耗时、耗钱和劳动力密集型的工作,“计算机辅助药物设计”的引入对整个研发过程都有一定的“辅助”,有时候甚至起到推动药物研发或者决定药物研发成败的关键因素。感兴趣的读者可以阅读Tanaji T. Talele等人近期的综述(Current Topics in 3
计算机辅助药物设计 朱瑞新着-- 计算机辅助药物设计(Ⅰ)--基本方法原理概要与实践详解
Medicinal Chemistry, 2010, 10, 127-141),其中对1、Captopril (Capoten?, Bristol
Myers-Squibb),2、Dorzolamide (Trusopt?, Merck),3、Saquinavir (Invirase?, Hoffmann-La Roche),4、Zanamivir (Relenza?, Gilead Sciences),5、Oseltamivir (Tamiflu?, Gilead Sciences),
6、Aliskiren (Tekturna?, Novartis),7、Boceprevir (Schering-Plough),8、Nolatrexed
dihydrochloride (Thymitaq?, Agouron),9、TMI-005,10、LY-517717 (Lilly/Protherics),11、Rupintrivir (AG7088, Agouron),和12、NVP-AUY922 (Novartis)等12个利用“计算机辅助药物设计”方法发现或者优化并得到美国食品安全局批准的药物进行了介绍。[www.61k.com]读者还可以进一步阅读德国著名药物学家Hugo Kubinyi于2006年撰写的专著(Computer Applications in
Pharmaceutical Research and Development, Ekins, S. Ed. Wiley-Interscience, NY,2006年),从而获得对“计算机辅助药物设计”应用的更多自信。
这里特别要提醒读者的是,药物研究是个非常复杂,并且风险很高的事情,本书仅仅是为了方便读者的理解,才将“计算机辅助药物设计”研究分解成如上图1-2所示的七大研究方向。在实际研究中,往往需要综合运用上述研究方向的多个或全部核心技术。
例如在对药物进行高通量虚拟筛选,往往需要进行如下步骤过滤(摘自www.kubinyi.de):
1、 是否有书写错误等(Garbage filter);
2、 是否有类药性(Druglike / Non-druglike);
3、 是否达到一定的生物利用度(Bioavailability):定量构效关系和药效团模型;
:定量构效关系和药效团模型; 4、 是否具有细胞毒性(Cytotoxicity)
:定量构效关系、药5、 是否抑制hERG钾离子通道(hERG channel inhibition)
效团模型和分子对接;
6、 是否与下列常见抗靶标有作用(Antitargets):
(1)?1a (orthostatic hypotension);
(2)D2 (extrapyramidal syndrome);
(3)5-HT2c (obesity);
(4)musc. M1 (hallucinations, memory);
定量构效关系、药效团模型、同源模建和分子对接;
7、 是否对P450代谢酶有抑制作用(CYP inhibition (3A4, 2C9, 2D6)):定量构
效关系、药效团模型和分子对接。
在这样一个通用的药物虚拟高通量筛选过程中,就需要直接的综合运用大分子结构预 4
计算机辅助药物设计 朱瑞新着-- 计算机辅助药物设计(Ⅰ)--基本方法原理概要与实践详解
测、定量构效关系、药效团模型和分子对接四种方法;并且需要间接的运用同源模建、分子库设计和动力学模拟三种方法。(www.61k.com)
本节最后简要地对“计算机辅助药物设计”所用到的方法基础进行概括。更为详细的参考请阅读中国科学院上海有机化学研究所陈敏伯研究员近期撰写的《计算化学:从理论化学到分子模拟》(科学出版社,2009年版)。需要强调的是:本书无意对“计算机辅助药物设计”方法中涉及到的纯粹数学方法作深入介绍,作者认为对这些内容的过多讲解往往分散读者的注意力,最终导致读者不能很好的从整体上了解和掌握“计算机辅助药物设计”相关知识。
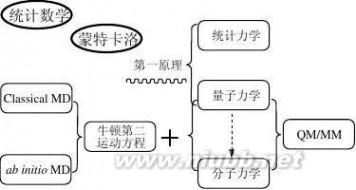
图1-3、“计算机辅助药物设计”的方法基础
如上图(图1-3)所示,同其它自然科学一样,“计算机辅助药物设计”的基础理论也是建立在“数学”和“物理”这两个最基础的学科之上。
1、 数学
除了常规的数学知识外,“计算机辅助药物设计”的多个核心技术对统计数学和蒙特卡洛十分偏爱,尤其是定量构效关系研究中,各种统计数学方法的引入让该技术从诞生至今仍受到广泛的重视(详见本书第四章),而在构象优化等问题上,蒙特卡洛算法一直是讨论的热点。
2、 物理
“事实上,量子力学规律不仅支配着微观世界,而且也支配着宏观世界,可以说全部物理学都是量子物理学的。已被长期实践证明的描述宏观现象的经典力学规律,实质上不过是量子力学规律的一个近似。”
简单来讲,“量子力学”是研究微观粒子各种属性的物理方法;“统计力学”则是将粒子们的微观属性同它的宏观表现关联起来的物理方法;而一切基于“量子力学”和“统计力学”演绎而得到的方法就是所谓的“第一原理”或者“从头算原理”方法。量子力学发展至今,尚未得出矛盾的结论,这样一来,便有了“从第一原理出发就可以解释一切现象”的观点。 5
计算机辅助药物设计 朱瑞新着-- 计算机辅助药物设计(Ⅰ)--基本方法原理概要与实践详解
量子力学的发展,根据是否直接计算电子的波函数而形成了两大类研究方法:(1)、波函数和(2)、密度泛函。(www.61k.com]1998年诺贝尔化学奖就是分别授予了这两类方法的代表人物John A. Pople和Walter Kohn。
可是,令人沮丧的是,即使选用计算量较低的密度泛函方法,对于生物大分子来讲仍然是不大现实的。而前面提到,作为药物受体的典型代表之一的生物大分子,在药物设计研究中显然是无法避免的。
于是类似于AM1、PM3和MNDO这些半经验的“从头算”方法不断地出现并一度成为该领域的通用方法。概括来讲,这些半经验方法都属于波函数派系,利用一定的实验数据信息拟合成参数简化原来“第一原理”相关方程中的部分项,比如利用高斯函数替代“原子实”的时候。
与此同时,LS-DFT(Linear-Scaling Density Functional Theory, 线性密度泛函法)等基于简化“第一原理”中密度泛函计算模型的方法也得到发展。
即便是使用半经验量化和线性密度泛函法,对于生物大分子的研究仍然不是很现实,在这样的基础上,大家不得转而使用对量子力学完全参数化的分子力学方法,由于分子力学方法中最关键的是各种简化的分子力场参数,因此这种方法经常也叫分子力场法。该方法将分子的能量可以近似看作构成分子的各个原子的空间坐标的函数,描述这种分子能量和分子结构之间关系的就是分子力场函数, 而分子力场函数来自实验结果的经验公式。
分子力场方法的使用使得大分子体系的研究得到了迅速发展,分子力场方法的计算量相对量子力学方法而言要小数十倍,而且在适当的精度内,分子力场方法得出的结果与量子力学计算结果相差无几。但是要注意的是:分子力场方法在研究分子中含有键的生成或断裂时是不适用的。这是分子力学方法的先天不足之处。
药物设计中经常会要研究大分子和小分子相互作用的复合体系。为了兼顾小分子与大分子相互作用界面的精度和计算量大小,将量子力学(QM)和分子力学(MM)杂合在一起用于体系的研究,即在相互作用界面采用精度高的量子力学进行研究,而在远离相互作用界面的地方(主要是大分子的部分残基)使用计算量小的分子力学进行研究。这样就诞生了QM/MM方法。
分子动力学、随机动力学和蒙特卡洛方法都可以用于计算体系的热力学量和其他宏观性质,分子动力学是以各种力学(包括经典力学和量子力学)和61阅读络类方法再试(当然一般在实际的药物设计中,并不建议做到这一步)。[www.61k.com)
比如,此例中,我们首选进行剔除Outlier操作。
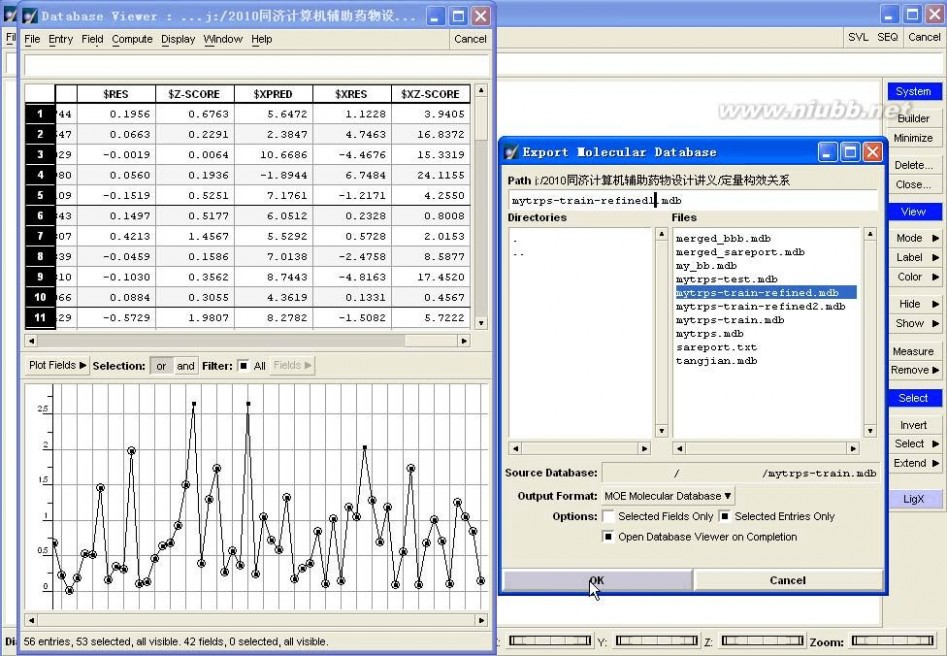
图4-15、剔除Outlier
具体操作:在训练集数据库中,首先DBV|Display,并勾上Plot,在Plot Fields中勾上$Z-SCORE,然后选择Z-SCORE中小于2的样本(剔除Outlier),单独保存为“mytrps-train-refined1.mdb”数据库,如图4-15所示。
然后在新产生的训练集的基础上重复步骤(4)。
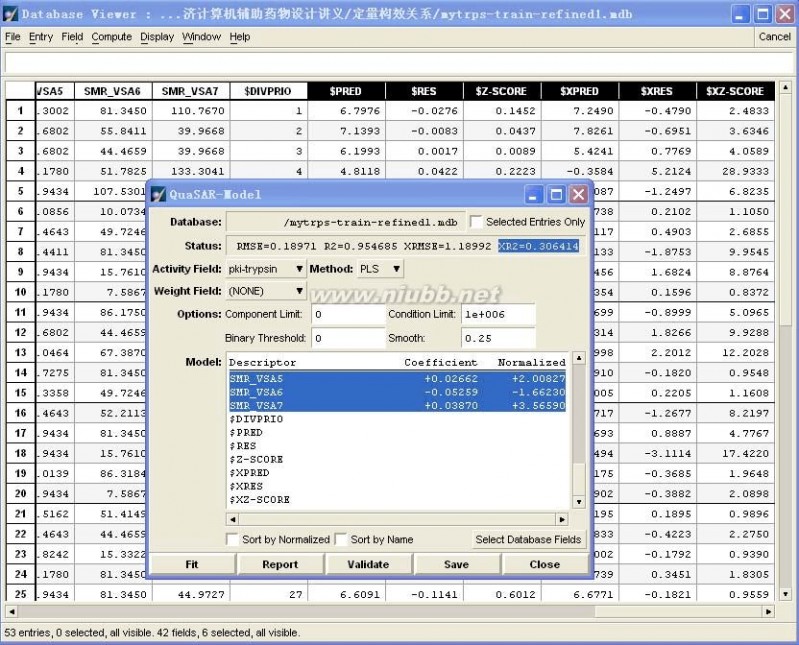
图4-16、第二次训练集自身交叉验证的结果
80
四 : 计算机辅助药物设计概论 1-1

计算机辅助药物设计 计算机辅助药物设计概论 1-1

计算机辅助药物设计 计算机辅助药物设计概论 1-1

计算机辅助药物设计 计算机辅助药物设计概论 1-1

计算机辅助药物设计 计算机辅助药物设计概论 1-1

计算机辅助药物设计 计算机辅助药物设计概论 1-1

计算机辅助药物设计 计算机辅助药物设计概论 1-1

计算机辅助药物设计 计算机辅助药物设计概论 1-1
计算机辅助药物设计 计算机辅助药物设计概论 1-1
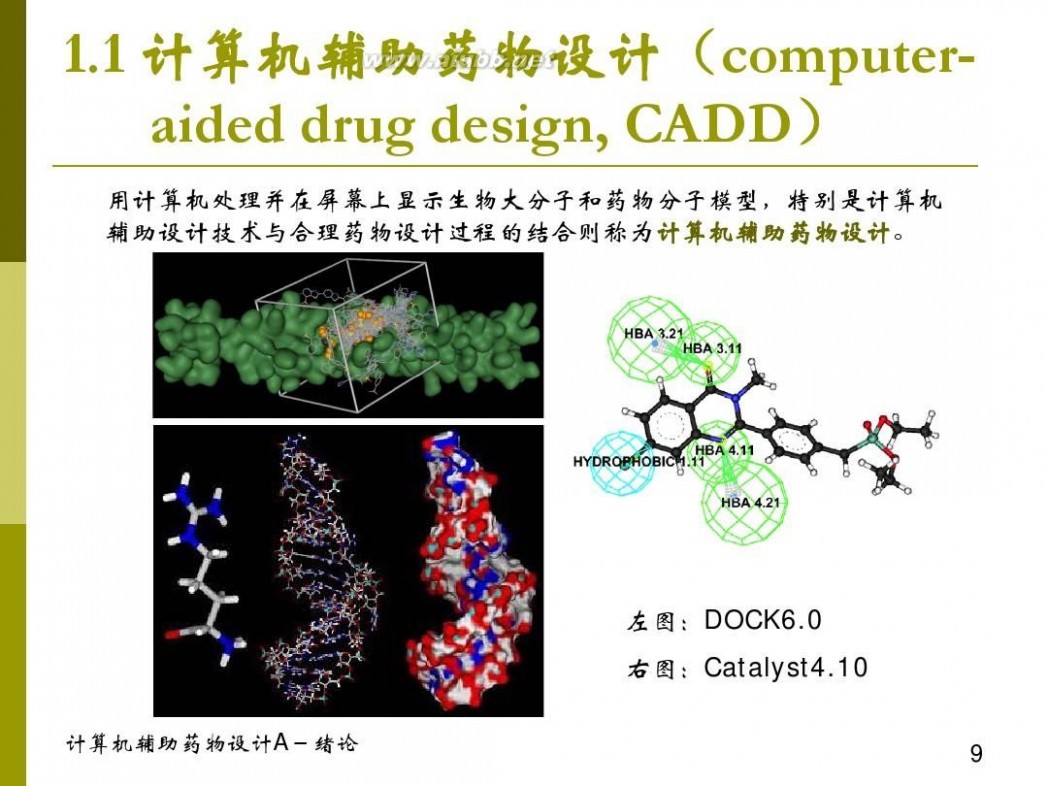
计算机辅助药物设计 计算机辅助药物设计概论 1-1

计算机辅助药物设计 计算机辅助药物设计概论 1-1

计算机辅助药物设计 计算机辅助药物设计概论 1-1
计算机辅助药物设计 计算机辅助药物设计概论 1-1
计算机辅助药物设计 计算机辅助药物设计概论 1-1

计算机辅助药物设计 计算机辅助药物设计概论 1-1
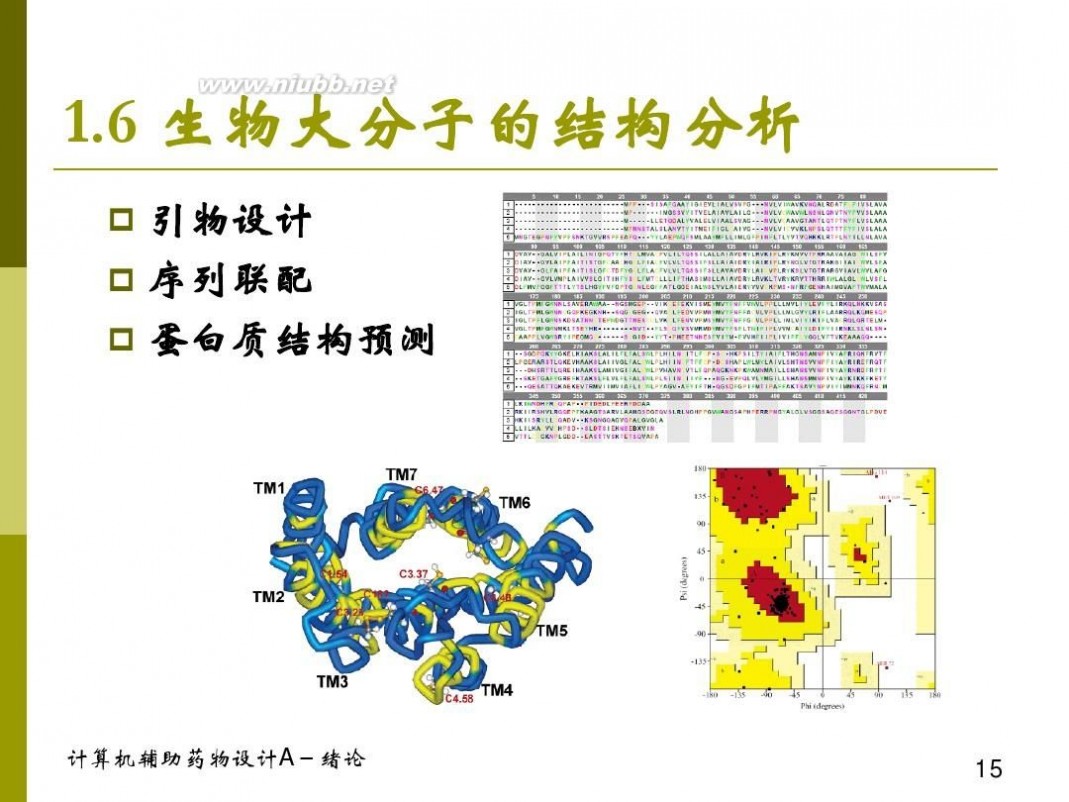
计算机辅助药物设计 计算机辅助药物设计概论 1-1

计算机辅助药物设计 计算机辅助药物设计概论 1-1
计算机辅助药物设计 计算机辅助药物设计概论 1-1

计算机辅助药物设计 计算机辅助药物设计概论 1-1

计算机辅助药物设计 计算机辅助药物设计概论 1-1
计算机辅助药物设计 计算机辅助药物设计概论 1-1
计算机辅助药物设计 计算机辅助药物设计概论 1-1
计算机辅助药物设计 计算机辅助药物设计概论 1-1

计算机辅助药物设计 计算机辅助药物设计概论 1-1
计算机辅助药物设计 计算机辅助药物设计概论 1-1

计算机辅助药物设计 计算机辅助药物设计概论 1-1

计算机辅助药物设计 计算机辅助药物设计概论 1-1 61阅读请您转载分享:

计算机辅助药物设计 计算机辅助药物设计概论 1-1
计算机辅助药物设计 计算机辅助药物设计概论 1-1

61阅读请您转载分享:
本文标题:计算机辅助药物设计-朱瑞新着-- 计算机辅助药物设计(Ⅰ)--基本方法原理61阅读| 精彩专题| 最新文章| 热门文章| 苏ICP备13036349号-1