一 : 色素失禁症研究进展
【摘要】 色素失禁症(Incontinentia pigmenti,IP)是一种X连锁显性遗传性疾病,其主要症状表现为患儿出生时或生后两周皮肤出现红斑及水疱,继水疱后在相同部位出现疣状损害,之后表现为奇特的网状色素沉着。IP还伴有其他系统症状。本病大多愈后良好。本文就该病发病机制、症状及诊断、鉴别诊断作一综述。
【关键词】 色素失禁症; 红斑; 疣状损害; 网状色素沉着
色素失禁症(Incontinentia pigmenti,IP)由Bloch于1926年首次报道,Sulzberger 于1982年对本病做了进一步描述,故又称Bloch-Sulzberger综合征。本病是一种罕见的系统性疾病,女孩与男孩发病比为20:1。该病是一种X连锁显性遗传性疾病,女性发病倾向更为显著。因为异常基因在性染色体上,女性因存在于另一个X染色体的正常基因可将其覆盖,故症状表现不甚严重,而男性仅有一个X染色体,因而病变表现严重,多半于胎儿期死亡[1]。据报道极少数男孩患病可幸存,因其染色体核型为:47,XXY。
1 IP发病机制的研究
1.1 引起IP突变基因定位 目前证实定位于X染色体长臂的Xq11(IP1)和Xq28(IP2)突变引起。
1.2 引起IP的突变基因
1.2.1 NEMO基因 位于Xq28,即NEMO(nuclear factor kB essential modulator)基因,亦称IKKy(theY-Subunit of the inhibitor kBkinase)。其NEMOcDNA全长2035 bp,由10个外显子组成,NEMO区域有2个卷曲螺旋区(CC1和CC2),1个亮氨酸拉链区(LZ)和1个锌指区(ZF)[2-3]。Smahi等[1]和Aradhya等[4]分别对47例和357例IP患者NEMO基因分析发现有80%、90%患者表现为NEMO基因4-10外显子缺失突变,NEMO基因4-10外显子缺失突变,导致IP患者发病,仍有一部分IP患者发病病因不明。
1.2.2 ?NEMO基因 在同一X染色体,人类NEMO序列具有两个拷贝,其中一个为无功能的假基因 ?NEMO,只包含外显子3~10,22 kb大小。它在NEMO基因的端粒处,距离NEMO基因第10外显子约85 kb,与NEMO基因方向相反。翻转重复存在NEMO基因外显子10的下游35.5 kb大小的重复序列内,重复序列始于外显子3上游,止于外显子10下游[5]。假基因不表达,NEMO基因内共有序列NEMO 4~10缺失发生色素失禁病,假基因 ?NEMO基因内该片段缺失不导致发病。日本一个关于IP调查,调查对象10人,其中有一半人发现有基因重排,因此笔者认为NEMO基因重排频率在日本患者与其他国家患者相似[6]。
1.2.3 NEMO基因与G6PD基因重叠 NEMO位于X染色体独立基因组,在着丝点方向,部分NEMO基因与G6PD基因重叠,而两个重叠基因共用一个固定启动子区域(promoterB),这个区域具有双向调节性,G6PD基因改变还没有被发现[7-9]。但有专家认为G6PD基因改变造成IP男性患者致死[10-11]。
G6PD基因缺乏症是一种酶缺乏的X连锁性遗传性疾病,是一种常见的酶缺乏病,多累及男性,表现为食用蚕豆后出现非免疫性溶血性贫血,杂合子女性也可表现出G6PD基因缺乏症状。人类G6PD基因表现为多态性,几乎G6PD基因所有突变都是点突变,因此导致了氨基酸置换,目前尚未发现大片段G6PD基因突变。
2012年Fuscol等[12]第一次报道IP患者同时存在G6PD基因缺失,该患者无任何G6PD基因缺失典型表现。NEMO缺失引发该患者IP,而G6PD等位基因选择性的覆盖了G6PD缺失所引发的症状,因此该患者无G6PD基因缺失典型表现。
1.3 NEMO基因产物 NEMO基因产物即为NEMO或叫IKKγ,是富含谷氨酰胺的48 000大小的蛋白质,它是IκB激酶复合物组成部分[13]。IκB激酶复合物是由IKKα,IKKβ 和NEMO组成,能激活NF- B信号通道[14]。其中IKKα、IKKβ为具有催化活性的亚单位。
NEMO是IкB激酶(IKK)基本调节亚单位,它编码IKB激酶复合物,这种IKB激酶复合物是核因子(NF)-κB激活过程中的关键酶,IKB激酶与细胞生长、活动、功能维持密切相关。(NF)-κB转录因子是一种和免疫及炎症反应相关的转录因子,能保护细胞免受TNF介导的凋亡。NEMO对于激活(NF)-κB转录因子和对抗肿瘤坏死因子诱导的细胞凋亡是至关重要[15]。NEMO基因突变致使(NF)-kB激活物发生改变,并使得细胞凋亡易感[16]。Sebban等[17]提出,NEMO角质缺陷容易受细菌的感染而破坏,并释放出细胞内容物,释放的细胞内容物能够介导炎性反应,如正常的NEMO细胞释放的干扰素TNF-α和白介素1β。文献[15]中报道的病例有巨大的疣状损伤,可能与NEMO角质缺陷增殖有关。笔者推测大量的疣状损伤可能是NEMO角质缺陷增殖的一种形式。
Thakur等[18]2011年报道了四个IP家庭,这四个家庭中的女儿都通过分子机制明确诊断了IP且都有典型的IP表现。这个分子机制是通过提取被影响的母亲和女儿的DNA,用Bardaro等[19]描述的PCR技术进行分析,确定了NEMO基因(1045 bp)外显子4-10缺陷,这种基因的缺失改变了mRNA第399个核苷酸之后的序列,从而导致翻译出的蛋白质缩短。这项技术还检测到IP共同重排序列,如在NEMO基因上外显子4-10的基因重排,还包括13个新突变单位[18]。由NEMO突变引起的IP患者占到全部患者的60%~80%[20]。在文献[18]中1号和4号家庭的母亲被确定有这段基因的缺陷,其他两个家庭的母亲正常。西方国家报道了大量的IP患者的研究,其中一些报道在我国也适用[21]。一般而言,中西方的NEMO突变频率是相似的,除了在女性患者身上发现的基因重排外,在男性患者身上这种重排都是致死性的,NEMO的突变包括69种不同的小突变,包括错位、移位、无意义密码子,剪切位点、突变等。
2 IP的症状
2.1 IP皮肤表现 IP皮肤表现临床分为四期:第一期为囊泡期,囊泡内多含嗜酸性粒细胞,亦称炎症反应期,约90%患者发病;第二期为疣状皮疹期,有红斑及水疱,排列成行,出生时即有或出生后2周内显著,常波及四肢和躯干,不侵犯面部,约70%患者发病,是继水疱后在相同部位出现的损害,这些损害通常1岁时消失,也可持续多年;第三期为色素沉着期,表现为奇特的网状色素沉着,约98%患者出现此期皮肤损害;第四期为萎缩期,此期表现为皮肤苍白,斑状萎缩,色素减少,以躯干部损害最显著,至成年期通常不易觉察,萎缩部位皮肤无毛发[1,22]。
2.2 IP神经系统改变表现 除了皮肤损害外,IP患者还有神经系统的表现,包括癫痫、小儿脑病、急性播散性脑脊髓炎、缺血性中风等。IP患者中有神经系统表现的占IP患者总数的30%[23]。大多神经症状表现在新生儿期,极少患者表现在青春期和成人期。MRI检查结果包括脑室周围和白质疾病、出血性疾病、胼胝体发育不全、脑萎缩和小脑发育不全[24]。
2.3 IP口腔及牙齿表现 Mini?等[25]调查的1993-2010年报道的IP患者中,其中有牙齿和口腔的异常占54.38%。所有牙齿和口腔异常的患者中男女比例无差异,牙齿异常常表现为牙齿的畸形、发育异常和出牙延迟。常见的口腔异常表现为唇腭裂和高颚弓[11]。牙齿及口腔的异常往往影响患者的生活质量。但不同于其他IP症状的是牙齿及口腔的异常能够被矫正。
2.4 眼部病变 眼部异常病例占25%~35%,其中严重受累约19%[26]。眼部的表现包括:斜视、视网膜血管的变化、视神经萎缩、小眼畸形和白内障等。早在1998年就有学者发现:IP患儿出生后眼底存在与早产儿视网膜病变、糖尿病、镰状细胞贫血等疾病相似的眼底斑点状血管缺血性改变,视网膜血管异常:动静脉吻合,周边区域低灌注,继发性新生血管形成,视网膜前纤维组织形成,晶状体后团块形成[27]。这提示临床医生对IP新生儿眼底检查及其治疗,有可能改变IP患者眼部异常的预后。
2.5 其他系统损害 其他系统损害还包括骨骼发育异常,如小头畸形、侏儒症、畸形足等。
3 诊断
目前IP的诊断依然依据1993年Landy和Donnai提出的国际公认的诊断标准[28],其主要内容如下: 有阳性家族史,主要标准如下:a典型皮疹史;b色素沉着;c皮肤毛发损害;d秃顶;e牙齿异常;f视网膜病变;g多次妊娠男胎流产证据。无阳性家族史,主要指标:a典型新生儿期的红斑、水疱,水疱内含嗜酸性粒细胞;b典型躯干部线状色素沉着;c皮肤线状萎缩或秃发。次要指标:a牙齿异常;b秃发;c指甲异常;d视网膜病变。临床诊断标准如下:无阳性家族史至少需要1条主要指标及1条次要指标支持诊断;有阳性家族史有1条临床标准即可诊断。
2013年Mini?等[29]建议将IP诊断标准进行调整。建议如下:主要标准在原有主要标准基础上增加IP患者皮损四个阶段中的任意一个,将多次妊娠男胎流产证据改为次要标准;次要标准在原有次要标准基础上增加了乳房及乳头异常、IP病理学诊断。嗜酸性粒细胞增多及不平衡X-染色体失活研究也可考虑作为IP诊断条件。
除了IP诊断的主要及次要标准外,IP典型IKBKG突变及家庭中相关成员IP诊断,都可考虑IP诊断;如果缺乏一级女性亲属IP诊断及IKBKG突变数据,至少有两个或两个以上主要标准及一个或一个以上次要标准来确诊散发IP病例;如果缺乏一级女性亲属IP诊断但却有IKBKG典型突变,任何一个主要或次要标准都可诊断IP;如果有一级女性亲属IP诊断证据,任何一个主要或至少两个次要标准都可诊断IP[29]。
当累积到中枢系统时可进行MRI和MRS检查,多数学者认为颅脑损害的原因是脑部血管闭锁、缺血和出血。MRI和MRS检查可有如下表现:(1)皮质和皮质下区白质异常短T1长T2信号;(2)脑白质内软化囊腔;(3)大、小脑单侧萎缩和/或胼胝体发育不全;(4)侧脑室扩大,室周白质T2WI高信号;(5)神经元标志物NO乙酰天门冬氨酸减少,乳酸水平增高[15]。利用基因检测诊断IP费用昂贵,不适合普遍投入临床应用。
4 鉴别诊断
本病根据病史及特征性皮肤表现诊断较容易,在疣状皮疹期应与下列疾病进行鉴别。
4.1 新生儿脓疱疮 好发于新生儿,起病急。基本损害为广泛分布的多发性大脓疱,尼氏征阳性,脓疱周围有红晕。疱壁薄,易破溃,破溃后成红色糜烂面。可伴有高热、畏寒等全身中毒症状,严重者可危及生命。脓液细菌培养可见金黄色葡萄球菌或溶血性链球菌生长。IP患儿一般无全身症状。
4.2 先天性大疱性表皮松解症 幼年发病者大多有家族史,皮损多见于四肢关节伸侧及其他易摩擦部位,水疱消退后有糜烂和萎缩,病情较重者可累及黏膜。组织病理表现为表皮下疱,浸润细胞少。色素失禁症极少累及黏膜。
4.3 幼年大疱性类天疱疮 发病年龄大多小于5岁,可以在出生后数周内出现,男孩多见。对称分布于颈部、胸腹部和四肢屈侧,亦可累及掌跖,黏膜损害比成人多见。反复发作3~4年可以自愈。组织病理检查可与色素失禁症鉴别。
4.4 Franceschetti-Jadasson综合征 该病色素呈网状分布,无牙齿异常和眼部损害。
5 结语
尽管基因调节体(NEMO)基因突变导致了IP已经明确,在IP发病过程中是否还有其他突变基因起作用有待于进一步研究。IP是否与G6PD基因有关及相关机制需进一步研究。IP除了上述描述症状外是否还有其他症状有待于进一步发现。参考文献
[1] Smahi A,Courtois G,Vabres P,et al.Genomic rearrangement in NEMO impairs NF-κB activation and is a cause of incontinentia pigmenti[J].Nature,2000,405(6785):466-472.
[2] Fusco F,Bardaro T,Fimiani G,et al.Molecular analysis of the genetic defect in a large cohort of IP patients and identification of novel NEMO mutations interfering with NF-κB activation[J].Hum Mol Genet,2004,13(16):1763-1773.
[3] Zonana J,Elder M E,Schneider L C,et al.A novel X-Linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO)[J].Am J Hum Genet,2000,67(6):1555-1562.
[4] Aradhya S,Woffendin H,Jakins T,et al.A recurrent deletion in the ubiquitously expressed NEMO (IKK-γ) gene accounts for the vast majority of incontinentia pigmenti mutations[J].Hum Mol Genet,2001,10(19):2171-2179.
[5] Aradhya S,Bardaro T,Galgóczy P,et al.Multiple pathogenic and benign genomic rearrangements occur at a 35 kb duplication involving the NEMO and LAGE2 genes[J].Hum Mol Genet,2001,10(22):2557-2567.
[6] Franzè A,Ferrante M I,Fusco F,et al.Molecular anatomy of the human glucose 6-phosphate dehydrogenase core promoter[J].FEBS Lett,1998,437(3):313-318.
[7] Galgóczy P,Rosenthal A,Platzer M.Human-mouse comparative sequence analysis of the NEMO gene reveals an alternative promoter within the neighboring G6PD gene[J].Gene,2001,271(1):93-98.
[8] Fusco F,Mercadante V,Miano M G,et al.Multiple regulatory regions and tissue-specific transcription initiation mediate the expression of NEMO/IKKγ gene[J].Gene,2006,15(383):99-107.
[9] Vulliamy T,Beutler E,Luzzatto L.Variants of glucose-6-phosphate dehydrogenase are due to missense mutations spread throughout the coding region of the gene[J].Hum Mutat,1993,2(3):159-167.
[10] Luzzatto L.Glucose 6-phosphate dehydrogenase deficiency:from genotype to phenotype[J].Haematologica,2006,91(10):1303-1306.
[11] Cappellini M D,Fiorelli G.Glucose-6-phosphate dehyd[www.61k.com)rogenase deficiency[J].The lancet,2008,371(9606):64-74.
[12] Fusco F,Paciolla M,Napolitano F,et al.Genomic architecture at the Incontinentia Pigmenti locus favours de novo pathological alleles through different mechanisms[J].Hum Mol Genet,2012,21(6):1260-1271.
[13] Carrascosa R M C,Ruiz C R,Medina M C,et al.Neonatal convulsions caused by incontinentia pigmenti with left opercular dysgenesia[J].Rev Neurol,2002,36(1):36-39.[14] Yoshikawa H,Uehara Y,Abe T,et al.One-month-old girl with incontinentia pigmenti[J].Neurology,1999,53(8):1633.
[15] Yoshida M,Oiso N,Kimura M,et al.Skin ulcer mimicking pyoderma gangrenosum in a patient with incontinentia pigmenti[J].The Journal of Dermatology,2011,38(10):1019-1021.
[16] Yamaoka S,Courtois G,Bessia C,et al.Complementation cloning of NEMO,a component of the IκB kinase complex essential for NF-κB activation[J].Cell,1998,93(7):1231-1240.
[17] Sebban H,Courtois G.NF-κB and inflammation in genetic disease[J].Biochem Pharmacol,2006,72(9):1153-1160.
[18] Thakur S,Puri R D,Kohli S,et al.Utility of molecular studies in incontinentia pigmenti patients[J].Indian J Med Res,2011,33(4):442.
[19] Bardaro T,Falco G,Sparago A,et al.Two cases of misinterpretation of molecular results in incontinentia pigmenti,and a PCR-based method to discriminate NEMO/IKKγ dene deletion[J].Hum Mutat,2003,21(1):8-11.
[20] O'Doherty M,Mc Creery K,Green A J,et al.Incontinentia pigmenti-ophthalmological observation of a series of cases and review of the literature[J].Br J Ophthalmol,2011,95(1):11-16.
[21] Hsiao P F,Lin S P,Chiang S S,et al.NEMO gene mutations in Chinese patients with incontinentia pigmenti[J].J Formos Med Assoc,2010,109(3):192-200.
[22] Berlin A L,Paller A S,Chan L S.Incontinentia pigmenti:a review and update on the molecular basis of pathophysiology[J].J Am Acad Dermatol,2002,47(2):169-190.
[23] Carney Jr R G.Incontinentia pigmenti:a world statistical analysis[J].Arch Dermatol,1976,112(4):535.
[24] Meuwissen M E C,Mancini G.Neurological findings in incontinentia pigmenti;a review[J].European Journal of Medical Genetics,2012,55(5):323-331.
[25] Mini? S,Trpinac D,Gabriel H,et al.Dental and oral anomalies in incontinentia pigmenti:a systematic review[J].Clin Oral Invest,2013,17(1):1-8.
[26]李莉,宋国维,徐放生,等.色素失禁症15例临床研究[J].中国实用儿科杂志,2005,20(8):472-474.
[27] Goldberg M F.Macular vasculopathy and its evolution in incontinentia pigmenti[J].Ophthalmic Genet,1998,19(3):141-148.
[28] Hadj-Rabia S,Froidevaux D,Bodak N,et al.Clinical study of 40 cases of incontinentia pigmenti[J].Arch Demato,2003,139(9):1163-1170.
[29] Mini? S,Trpinac D,Obradovi? M.Incontinentia pigmenti diagnostic criteria update[J].Clin Genet,2013,44(6):1-3.
(收稿日期:2013-11-08) (本文编辑:欧丽)
二 : 色素失禁症

| 疾病名称(英文) | incontinentia pigmenti |
| 拚音 | SESUSHIJINZHENG |
| 别名 | |
| 西医疾病分类代码 | 遗传性疾病,皮肤科疾病, |
| 中医疾病分类代码 | |
| 西医病名定义 | 色素失禁症(incontinentia pigmenti)是一种以水疱、疣状丘疹和色素沉着为主要表现的遗传性皮肤病。 |
| 中医释名 | |
| 西医病因 | 可能为X联显性遗传,女性的异常基因只位于两个性染色体"XX"中的一个,所以病变不太严重,而男性的异常染色体基因则位于其单个性染色体"x"上,所以病变严重,常在胎儿期死亡。因此,临床上多见于女婴。 |
| 中医病因 | |
| 季节 | |
| 地区 | |
| 人群 | |
| 强度与传播 | |
| 发病率 | |
| 发病机理 | |
| 中医病机 | |
| 病理 | |
| 病理生理 | |
| 中医诊断标准 | |
| 中医诊断 | |
| 西医诊断标准 | |
| 西医诊断依据 | 根据特征性的喷洒状色素沉着,诊断不难。 |
| 发病 | |
| 病史 | |
| 症状 | |
| 体征 | 皮损发展可分为三期:第一期,在婴儿出生后1周左右于躯干、四肢发生红斑、水疱、风团样皮损,常排列成行。约经2月左右,转入第二期,表现为线状的疣状丘疹,稍带炎症。此期也可持续2个月左右,进入第三期,主要表现为色素性斑疹,形态特殊,犹如涡轮状或水泼状。数年后皮损逐渐减少甚至完全消失。患者一般情况良好,但少数病例可伴有其他系统症状,如智力迟钝、癫痫、白内障、斜视、视神经萎缩、渗出性视网膜脉络膜炎、心脏病、并指症、多肋骨、偏侧萎缩、腿和臀部缩短、出牙延迟、牙齿变尖、畸形或脱落,以及假性秃发、甲萎缩、掌跖多汗等。 |
| 体检 | |
| 电诊断 | |
| 影像诊断 | |
| 实验室诊断 | |
| 血液 | |
| 尿 | |
| 粪便 | |
| 脑脊液 | |
| 其他诊断 | |
| 免疫学 | |
| 组织学检验 | |
| 西医鉴别诊断 | 本症应与大疱表皮松解症及儿童期的大疱性类天疱疮鉴别,后两种病极少有本病所见的特征性色素沉着形态。与Franceschetti Jadassohn综合征的区别是后者的色素沉着呈网状,且无牙齿异常及眼部病变。 |
| 中医类证鉴别 | |
| 疗效评定标准 | |
| 预后 | 病变常在2岁时逐渐消退。至成人后,除原有的一些并发症外,无任何主观不适。 |
| 并发症 | |
| 西医治疗 | 本症不需特别治疗。 |
| 中医治疗 | |
| 中药 | |
| 针灸 | |
| 推拿按摩 | |
| 中西医结合治疗 | |
| 护理 | |
| 康复 | |
| 预防 | |
| 历史考证 |
三 : 新生儿色素失禁症临床特点分析

!1711!
果均示正常。临床上做了一系列动态观察,无1例符合ST??T动态演变,且在短期内恢复正常。其发生机制为:脑水肿颅压升高,致脑内血液循环障碍,累及下丘脑和脑干网状结构的心血管调节中枢,是致心电图异常的原因;也有认为,投射纤维与联合纤维激惹脑干诸核,导致交感神经兴奋性增高,儿茶酚胺合成分泌增多,致冠状动脉收缩和痉挛,致使出现ST段异常改变。中枢神经损伤后,通过反射儿茶酚胺在心肌的积聚,心肌广泛受损,影响心肌复极,延长复极完成时间,致使ST??T产生一系列改变。大部分属于??脑型心电图??改变,多呈一过性ST段抬高、异常J波、Q??T间期延长,并伴随恶性室性心律失常,应高度重视,防止出现猝死的危险。
脑源性心电图改变的发生机制是多方面的,如下丘脑植物神经功能紊乱、电解质紊乱或血中肾上腺素浓度升高有关,但多数作者强调植物神经功能障碍,尤其是丘脑受到损害或缺血是引起心电图变
化的主要原因。脑出血患者因颅压增高,均有不同程度呕吐,在使用脱水剂过程中很容易产生低钾、低镁血症。镁是许多酶的重要活化剂,具有兴奋心肌
作用,用镁以后可降低心律失常的发生率,镁能够阻滞自主神经,从而缓和了肾上腺素系统对心肌的激惹性,为此对于脑源性心律失常在应用脱水剂时除注意补钾外尚注意补镁[5]。
参考文献:
[1] 宋高英.脑梗死、脑出血患者心电图异常改变193例分析[J].实
用全科医学,2003,2(1):125??126.
[2] 卢喜烈.现代心电图诊断大全[M].北京:科学技术出版社,1996:9??25.
[3] 徐飞洪,陈武.118例脑梗死、脑出血患者心电图变化分析[J].
心血管康复医学杂志,2002,8(4):371.
[4] 朱力华,方炳森,张文旎.心电图精解[M].天津:科学技术出版
社,2004:128??129.
[5] 张荣.脑出血后尖端扭转型室性心动过速[J].临床心电学杂志,
2000,8:151.
收稿日期:2010??05??19 修回日期:2010??08??05 编辑:杜媛鲲
新生儿色素失禁症临床特点分析
郝丽红,荣丽英,麻庆荣,徐 莹
(天津市儿童医院新生儿内科,天津300074)
关键词:遗传性疾病,先天性;疹;体征和症状中图分类号:R758.5
文献标识码:B
文章编号:1004??583X(2010)19??1711??02
或类似病史。全部病例母亲在孕期无服用药物史。诊断依据:主要是依据1993年Landy和Donnai[1]提出的标准,新生儿期即出现皮肤红斑、疱疹、结节和疣状增生物;疱疹及疣状增生物合并出现;广泛播散的不规则泼墨状或涡轮状的色素沉着;部分有类似家族史。同时临床排除新生儿毒性红斑、脓疱疹、大疱性表皮松解症等疾病。本组5例中4例根据典型临床表现诊断,1例根据临床表现和皮肤活检病理结果诊断。
1.2 临床特征 5例患儿均有皮肤受累,3例宫内发病,出生时即有全身皮疹,另2例分别于生后6天和24天出现皮疹,5例患者中有3例于新生儿期出现惊厥发作,其中1例是以惊厥为其首发症状。5例患儿眼底检查均未发现异常。皮疹特征:2例有典型的皮疹过程:红斑水泡、疣状增生和色素沉着。皮疹初为疱疹样,内有淡黄色透明或半透明样液体,散在于躯干和四肢,疱疹周围皮肤红,部分融合成片,部分疱疹破溃有渗出,很快便形成疣状增生,高于皮肤,触感较硬(图1a~b),最后留有色素沉着,但各期色素失禁症(incontinentiapigmenti,IP)又称为
Bloch??Sulzbergersyndrome,是一种少见的X染色体连锁显性遗传性疾病,该症在新生儿期多数患者以皮肤损害为首发症状,而这种皮肤改变常被误诊为脓疱疹、湿疹、大疱表皮松解症等,使其诊断延误,但因该症可导致神经系统损害、智力发育迟缓、眼、牙齿、毛发等多处发育异常,临床需提高对该病的认识,早期诊断。现将我院收治的5例IP报告如下。1 临床资料
1.1 病例选择 2005年10月至2009年10月我院新生儿内科收治的IP患儿5例,均为女性,孕38~41周。顺产1例,剖宫产4例。出生体质量2.5~4.0kg。入院时间为2小时至27天,平均6天。其中2例有家族史(1例其母在婴幼儿期皮肤有色素沉着斑,后逐渐消退,另1例患者有一12岁姐姐反复皮,
!1712!
肢和躯干明显色素沉着,呈涡轮状。但5例患者皮疹形状不一,涡轮状、条索状或花纹状均有。1.3 实验室及病理检查 5例患儿血常规、C反应蛋白、血培养检查均阴性,宫内感染检查(巨细胞病毒PCR、弓形虫PCR、风疹病毒抗体、单纯疱疹病毒抗体、梅毒抗体)均阴性。血1,3??葡聚糖(-)。4例患儿疱液培养阴性。3例发生惊厥的患儿脑脊液检查未见异常,脑电图检查阴性,其中2例头CT检查未见异常,1例有脑室扩大。仅1例患儿行皮肤病理检查,提示表皮基底层黑色素细胞减少,真皮层黑色素细胞侵润,棘细胞增生(图2)。2 讨 论
IP为X染色体显性遗传,因男性仅一条X染色体,病情严重,常在胎儿期死亡,因此临床上绝大多数为女性患者。男性幸存者常伴有其他染色体畸形[2],而女性发病临床症状表现多样[3]。本组5例患儿均为女性。国外报道整体发病率在女孩为1/50000,且半数以上病例有家族史。
据报道IP的患者中有60%~85%在新生儿期即会发病[4]。该症在新生儿期主要表现为皮肤损害,半数以上在宫内或出生时即会出现皮肤损害。故对于出生时即有皮疹的女孩一定要考虑到有本症的可能。皮肤损害一般分为4期。第1期为红斑水泡期,皮肤可见明显疱疹,泡内多含嗜酸性粒细胞,约90%患者发生;第2期为疣状增生期,约70%发生;第3期为色素沉着期,约98%患者出现此期皮肤损害;第4期为萎缩期,此期表现为皮肤斑状萎缩,色素减少,萎缩部位皮肤无毛发。在新生儿期神经系统损害发生率亦较高,其中以惊厥发作最为常见,曾报道1例以癫痫持续状态为首发症状者,经抗癫痫治疗后,其痫样发作仍很难控制。本组5例患者中就有3例出现惊厥发作,其中1例是以惊厥为首发症状,加用苯巴比妥后症状控制良好。目前中枢神经系统的致病机制尚不十分清楚,发病的可能机制是炎症和血管源性破坏性机制。神经系统损害还可表现为智力运动发育落后、脑发育不良、偏瘫、小脑共济失调等。这些症状一般在生后1年内出现。有随访发现学龄期可出现注意力不集中或学习困难,所以有必要对患者进行神经系统结构和功能的动态评估。
瑞典学者研究指出[8],IP患者可有77%出现眼部受累,其中严重受累达47%。有报道说[9],这种眼[4]
[7]
[6]
[5]
膜病变类似的缺血性血管病变,继发性新生血管形成,形成晶体后团块,所以对于在新生儿期出现症状的IP患儿必须早期进行眼底检查治疗及随访,随访时间应为3年[8??9],这样有可能改变眼睛的预后。目前研究证实IP患者的基因位于Xq28,即NEMO(nuclearfactorkBessentialmodulator)基因,亦称IKKy。NEMO基因改变导致核因子??B失活,使得肿瘤坏死因子 诱导的细胞凋亡亢进。国外发现的基因改变类型中有90%为NEMO基因内共有序列NEMO!4~10缺失,只有10%为微小突变[12],而国内的研究也证实了这一点[13]。
目前认为IP的远期预后取决于是否合并其他系统受累,尤其是神经系统和眼部,出现症状后治疗相对困难,预后不良。对于新生儿期的IP重在早期诊断和随访。(本文图见封二)
参考文献:
[1] LandySJ,DonnaiD.Incontinentiapigmenti(Bloch??Sulzberger
syndrome)[J].JMedGenet,1993,30(1):53??59.
[2] 孙东信.色素失禁症38例分析[J].中华皮肤科杂志,1996,29
(1):15??17.[3] Hadj??RabiaS,FroidevauxD,BodakN,etal.Clinicalstudyof
40casesofincontinentiapigmeti[J].ArchDemato,2003,139(9):1163??1170.
[4] 李莉,宋国维,徐放生,等.色素失禁症15例临床研究[J].中国
实用儿科杂志,2005,20(8):472??474.
[5] BerlinAl,PallerAS,ChanLS.Incontinentiapigmenti:areviewandupdateonthemolecularbasisofpathophysiology[J].JAm
AcadDermatol,2002,47(2):169??190.
[6] 杨斌,徐广珍.儿童色素失禁症临床特点研究[J].中国优生与
遗传杂志,2009,17(3):126??127.
[7] HennelSJ,EkertPG,VolpeJJ,etal.Insightsintothe
pathogenesisofcerebrallesionsinincontinentiapigmenti[J].PediatrNeurol,2003,29(2):148??150.
[8] HolmstromG,ThotenK.Ocularmanifestationsofincontinentiapigmenti[J].ActaOphthalmolScand,2000,78
(3):348??353.
[9] GoldbergMF.Macularvasculopathyanditsevolutionin
incontinentiapigmenti[J].OphthalmicGenet,1998,19(3):141??148.
[10] JinDY,JeangKT.Isolationoffull??lengthcDNA
and
chromosomallocalizationofhumanNF??kBmodulatorNEMO
toXq28[J].JBiomedSci,1999,6(2):115??120.
[11] MakrisC,RobertsJL,KarinM.Thecarboxyl??terminalregion
ofIkappaBkinasegamma(IKK??gamma)isrequiredforfullIKKactivation[J].MolCellBiol,2002,22(18):6573??6581.
[12] AradhyaS,WoffendinH,JakinsT,etal.Arecurrentdeletion
intheubiquitouslyexpressedNEMO(IKK??gamma)geneaccountsforthevastmajorityofincontinentiapigmentimutations[J].HumMolGenet,2001,10(19):2171??2179.
[13] 李莉,宋国维,杜军保,等.色素失禁症NEMO!4~10基因片段缺失的初步研究[J].中华儿科杂志,2005,43(2):89??92.
??10 :??26 编辑:[11][10]
新生儿色素失禁症临床特点分析
(正文见第1711页

)
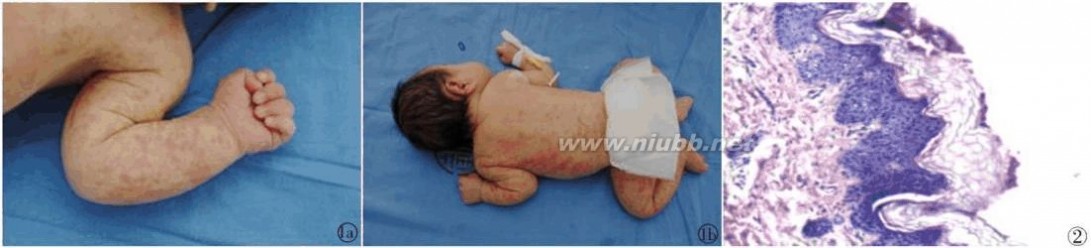
皮疹散在于躯干和四肢,呈红色,高于皮肤,触感较硬,呈涡轮样分布
图1 患儿皮疹临床表现 表皮基底层黑色素细胞减少,真皮层黑色素 细胞侵润,棘细胞增生 图2 皮疹病理学观察 HE?100
甲型H1N1流感危重症2例临床分析
(正文见第1724页

)
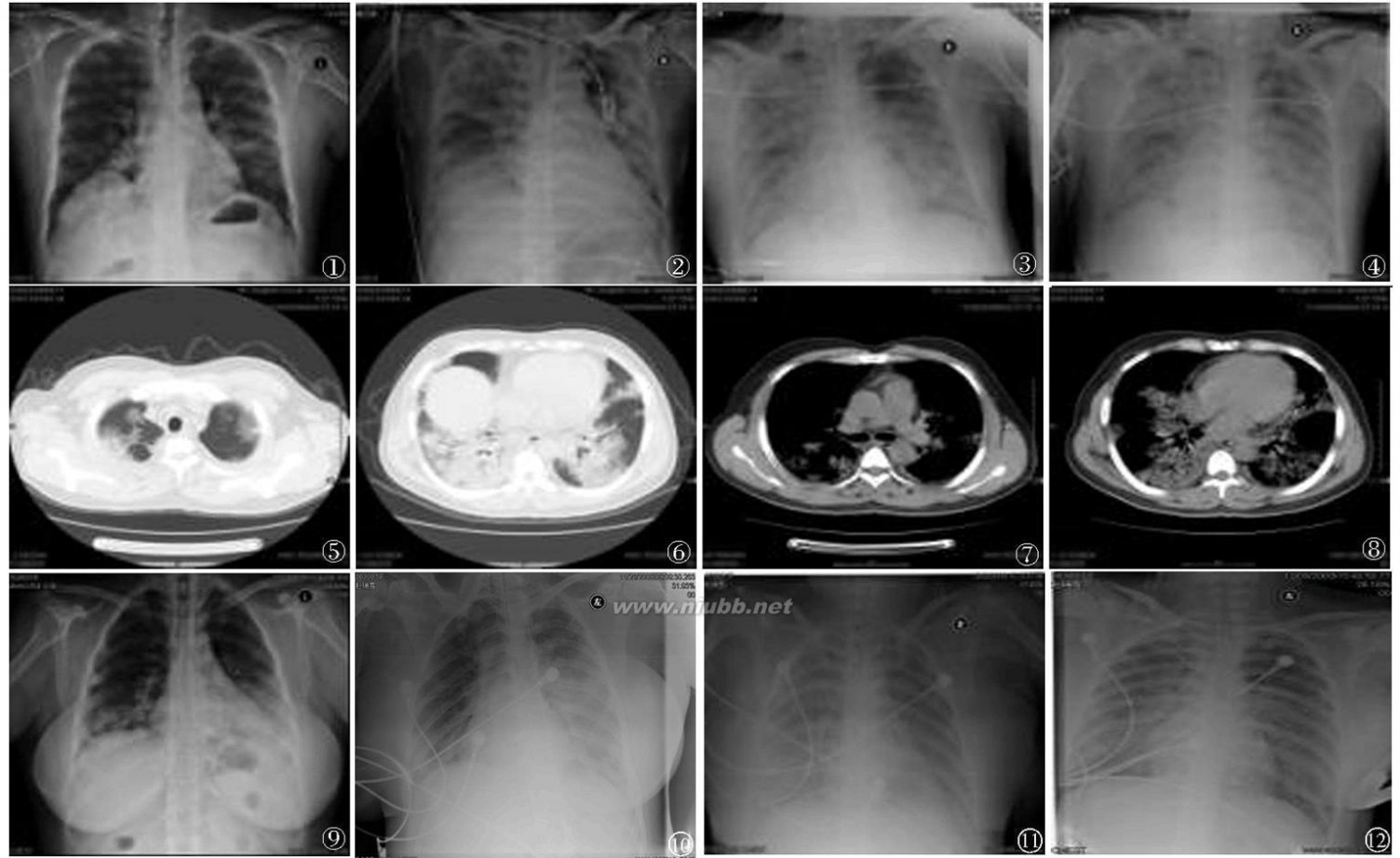
图1 例12009年11月21日胸部X线片(左肺肺门处出现斑片样增高密度影,右肺无明显异常);图2 例12009年11月24日胸部X线片(两肺斑片样高密度影明显增多,特别是右肺出现局限高密度影);图3 例12009年11月25日胸部X线片(两肺高密度影范围迅速扩大,更加致密,逐渐向两上肺野扩散);图4 例12009年11月26日胸部X线片(两肺弥漫性毛玻璃样改变);图5~8 例12009年11月23日胸部CT(双侧肺内多发斑片样阴影,伴有气管通气相);图9 例22009年11月24日胸部X线片(两下肺斑片样高密度影,尤其左肺明显);图10 例22009年11月26日胸部X线片(左肺高密度影较前明显扩大,右下肺高密度影较前增多);图11 例22009年12月2日胸部X线片(两肺弥漫性毛玻璃样改变);图12 例22009年12月8日胸部X线片(两肺弥漫性毛玻璃样改变,较前无改善)
十二指肠溃疡伴食物嵌顿1例
(正文见第1735页

)
图1 胃镜下嵌顿食物团块的十二指肠球部
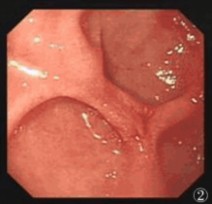
图2 胃镜下三枚较浅的假憩室
四 : 新生儿色素失禁症临床特点分析
!1711!
果均示正常。(www.61k.com)临床上做了一系列动态观察,无1例符合ST??T动态演变,且在短期内恢复正常。其发生机制为:脑水肿颅压升高,致脑内血液循环障碍,累及下丘脑和脑干网状结构的心血管调节中枢,是致心电图异常的原因;也有认为,投射纤维与联合纤维激惹脑干诸核,导致交感神经兴奋性增高,儿茶酚胺合成分泌增多,致冠状动脉收缩和痉挛,致使出现ST段异常改变。中枢神经损伤后,通过反射儿茶酚胺在心肌的积聚,心肌广泛受损,影响心肌复极,延长复极完成时间,致使ST??T产生一系列改变。大部分属于??脑型心电图??改变,多呈一过性ST段抬高、异常J波、Q??T间期延长,并伴随恶性室性心律失常,应高度重视,防止出现猝死的危险。
脑源性心电图改变的发生机制是多方面的,如下丘脑植物神经功能紊乱、电解质紊乱或血中肾上腺素浓度升高有关,但多数作者强调植物神经功能障碍,尤其是丘脑受到损害或缺血是引起心电图变
化的主要原因。脑出血患者因颅压增高,均有不同程度呕吐,在使用脱水剂过程中很容易产生低钾、低镁血症。镁是许多酶的重要活化剂,具有兴奋心肌
作用,用镁以后可降低心律失常的发生率,镁能够阻滞自主神经,从而缓和了肾上腺素系统对心肌的激惹性,为此对于脑源性心律失常在应用脱水剂时除注意补钾外尚注意补镁[5]。
参考文献:
[1] 宋高英.脑梗死、脑出血患者心电图异常改变193例分析[J].实
用全科医学,2003,2(1):125??126.
[2] 卢喜烈.现代心电图诊断大全[M].北京:科学技术出版社,1996:9??25.
[3] 徐飞洪,陈武.118例脑梗死、脑出血患者心电图变化分析[J].
心血管康复医学杂志,2002,8(4):371.
[4] 朱力华,方炳森,张文旎.心电图精解[M].天津:科学技术出版
社,2004:128??129.
[5] 张荣.脑出血后尖端扭转型室性心动过速[J].临床心电学杂志,
2000,8:151.
收稿日期:2010??05??19 修回日期:2010??08??05 编辑:杜媛鲲
新生儿色素失禁症临床特点分析
郝丽红,荣丽英,麻庆荣,徐 莹
(天津市儿童医院新生儿内科,天津300074)
关键词:遗传性疾病,先天性;疹;体征和症状中图分类号:R758.5
文献标识码:B
文章编号:1004??583X(2010)19??1711??02
或类似病史。全部病例母亲在孕期无服用药物史。诊断依据:主要是依据1993年Landy和Donnai[1]提出的标准,新生儿期即出现皮肤红斑、疱疹、结节和疣状增生物;疱疹及疣状增生物合并出现;广泛播散的不规则泼墨状或涡轮状的色素沉着;部分有类似家族史。同时临床排除新生儿毒性红斑、脓疱疹、大疱性表皮松解症等疾病。本组5例中4例根据典型临床表现诊断,1例根据临床表现和皮肤活检病理结果诊断。
1.2 临床特征 5例患儿均有皮肤受累,3例宫内发病,出生时即有全身皮疹,另2例分别于生后6天和24天出现皮疹,5例患者中有3例于新生儿期出现惊厥发作,其中1例是以惊厥为其首发症状。5例患儿眼底检查均未发现异常。皮疹特征:2例有典型的皮疹过程:红斑水泡、疣状增生和色素沉着。皮疹初为疱疹样,内有淡黄色透明或半透明样液体,散在于躯干和四肢,疱疹周围皮肤红,部分融合成片,部分疱疹破溃有渗出,很快便形成疣状增生,高于皮肤,触感较硬(图1a~b),最后留有色素沉着,但各期色素失禁症(incontinentiapigmenti,IP)又称为
Bloch??Sulzbergersyndrome,是一种少见的X染色体连锁显性遗传性疾病,该症在新生儿期多数患者以皮肤损害为首发症状,而这种皮肤改变常被误诊为脓疱疹、湿疹、大疱表皮松解症等,使其诊断延误,但因该症可导致神经系统损害、智力发育迟缓、眼、牙齿、毛发等多处发育异常,临床需提高对该病的认识,早期诊断。现将我院收治的5例IP报告如下。1 临床资料
1.1 病例选择 2005年10月至2009年10月我院新生儿内科收治的IP患儿5例,均为女性,孕38~41周。顺产1例,剖宫产4例。出生体质量2.5~4.0kg。入院时间为2小时至27天,平均6天。其中2例有家族史(1例其母在婴幼儿期皮肤有色素沉着斑,后逐渐消退,另1例患者有一12岁姐姐反复皮,
色素失禁症 新生儿色素失禁症临床特点分析
!1712!
肢和躯干明显色素沉着,呈涡轮状。[www.61k.com]但5例患者皮疹形状不一,涡轮状、条索状或花纹状均有。1.3 实验室及病理检查 5例患儿血常规、C反应蛋白、血培养检查均阴性,宫内感染检查(巨细胞病毒PCR、弓形虫PCR、风疹病毒抗体、单纯疱疹病毒抗体、梅毒抗体)均阴性。血1,3??葡聚糖(-)。4例患儿疱液培养阴性。3例发生惊厥的患儿脑脊液检查未见异常,脑电图检查阴性,其中2例头CT检查未见异常,1例有脑室扩大。仅1例患儿行皮肤病理检查,提示表皮基底层黑色素细胞减少,真皮层黑色素细胞侵润,棘细胞增生(图2)。2 讨 论
IP为X染色体显性遗传,因男性仅一条X染色体,病情严重,常在胎儿期死亡,因此临床上绝大多数为女性患者。男性幸存者常伴有其他染色体畸形[2],而女性发病临床症状表现多样[3]。本组5例患儿均为女性。国外报道整体发病率在女孩为1/50000,且半数以上病例有家族史。
据报道IP的患者中有60%~85%在新生儿期即会发病[4]。该症在新生儿期主要表现为皮肤损害,半数以上在宫内或出生时即会出现皮肤损害。故对于出生时即有皮疹的女孩一定要考虑到有本症的可能。皮肤损害一般分为4期。第1期为红斑水泡期,皮肤可见明显疱疹,泡内多含嗜酸性粒细胞,约90%患者发生;第2期为疣状增生期,约70%发生;第3期为色素沉着期,约98%患者出现此期皮肤损害;第4期为萎缩期,此期表现为皮肤斑状萎缩,色素减少,萎缩部位皮肤无毛发。在新生儿期神经系统损害发生率亦较高,其中以惊厥发作最为常见,曾报道1例以癫痫持续状态为首发症状者,经抗癫痫治疗后,其痫样发作仍很难控制。本组5例患者中就有3例出现惊厥发作,其中1例是以惊厥为首发症状,加用苯巴比妥后症状控制良好。目前中枢神经系统的致病机制尚不十分清楚,发病的可能机制是炎症和血管源性破坏性机制。神经系统损害还可表现为智力运动发育落后、脑发育不良、偏瘫、小脑共济失调等。这些症状一般在生后1年内出现。有随访发现学龄期可出现注意力不集中或学习困难,所以有必要对患者进行神经系统结构和功能的动态评估。
瑞典学者研究指出[8],IP患者可有77%出现眼部受累,其中严重受累达47%。有报道说[9],这种眼[4]
[7]
[6]
[5]
膜病变类似的缺血性血管病变,继发性新生血管形成,形成晶体后团块,所以对于在新生儿期出现症状的IP患儿必须早期进行眼底检查治疗及随访,随访时间应为3年[8??9],这样有可能改变眼睛的预后。目前研究证实IP患者的基因位于Xq28,即NEMO(nuclearfactorkBessentialmodulator)基因,亦称IKKy。NEMO基因改变导致核因子??B失活,使得肿瘤坏死因子 诱导的细胞凋亡亢进。国外发现的基因改变类型中有90%为NEMO基因内共有序列NEMO!4~10缺失,只有10%为微小突变[12],而国内的研究也证实了这一点[13]。
目前认为IP的远期预后取决于是否合并其他系统受累,尤其是神经系统和眼部,出现症状后治疗相对困难,预后不良。对于新生儿期的IP重在早期诊断和随访。(本文图见封二)
参考文献:
[1] LandySJ,DonnaiD.Incontinentiapigmenti(Bloch??Sulzberger
syndrome)[J].JMedGenet,1993,30(1):53??59.
[2] 孙东信.色素失禁症38例分析[J].中华皮肤科杂志,1996,29
(1):15??17.[3] Hadj??RabiaS,FroidevauxD,BodakN,etal.Clinicalstudyof
40casesofincontinentiapigmeti[J].ArchDemato,2003,139(9):1163??1170.
[4] 李莉,宋国维,徐放生,等.色素失禁症15例临床研究[J].中国
实用儿科杂志,2005,20(8):472??474.
[5] BerlinAl,PallerAS,ChanLS.Incontinentiapigmenti:areviewandupdateonthemolecularbasisofpathophysiology[J].JAm
AcadDermatol,2002,47(2):169??190.
[6] 杨斌,徐广珍.儿童色素失禁症临床特点研究[J].中国优生与
遗传杂志,2009,17(3):126??127.
[7] HennelSJ,EkertPG,VolpeJJ,etal.Insightsintothe
pathogenesisofcerebrallesionsinincontinentiapigmenti[J].PediatrNeurol,2003,29(2):148??150.
[8] HolmstromG,ThotenK.Ocularmanifestationsofincontinentiapigmenti[J].ActaOphthalmolScand,2000,78
(3):348??353.
[9] GoldbergMF.Macularvasculopathyanditsevolutionin
incontinentiapigmenti[J].OphthalmicGenet,1998,19(3):141??148.
[10] JinDY,JeangKT.Isolationoffull??lengthcDNA
and
chromosomallocalizationofhumanNF??kBmodulatorNEMO
toXq28[J].JBiomedSci,1999,6(2):115??120.
[11] MakrisC,RobertsJL,KarinM.Thecarboxyl??terminalregion
ofIkappaBkinasegamma(IKK??gamma)isrequiredforfullIKKactivation[J].MolCellBiol,2002,22(18):6573??6581.
[12] AradhyaS,WoffendinH,JakinsT,etal.Arecurrentdeletion
intheubiquitouslyexpressedNEMO(IKK??gamma)geneaccountsforthevastmajorityofincontinentiapigmentimutations[J].HumMolGenet,2001,10(19):2171??2179.
[13] 李莉,宋国维,杜军保,等.色素失禁症NEMO!4~10基因片段缺失的初步研究[J].中华儿科杂志,2005,43(2):89??92.
??10 :??26 编辑:[11][10]
色素失禁症 新生儿色素失禁症临床特点分析
新生儿色素失禁症临床特点分析
(正文见第1711页
)
皮疹散在于躯干和四肢,呈红色,高于皮肤,触感较硬,呈涡轮样分布
图1 患儿皮疹临床表现 表皮基底层黑色素细胞减少,真皮层黑色素 细胞侵润,棘细胞增生 图2 皮疹病理学观察 HE?100
甲型H1N1流感危重症2例临床分析
(正文见第1724页
)
图1 例12009年11月21日胸部X线片(左肺肺门处出现斑片样增高密度影,右肺无明显异常);图2 例12009年11月24日胸部X线片(两肺斑片样高密度影明显增多,特别是右肺出现局限高密度影);图3 例12009年11月25日胸部X线片(两肺高密度影范围迅速扩大,更加致密,逐渐向两上肺野扩散);图4 例12009年11月26日胸部X线片(两肺弥漫性毛玻璃样改变);图5~8 例12009年11月23日胸部CT(双侧肺内多发斑片样阴影,伴有气管通气相);图9 例22009年11月24日胸部X线片(两下肺斑片样高密度影,尤其左肺明显);图10 例22009年11月26日胸部X线片(左肺高密度影较前明显扩大,右下肺高密度影较前增多);图11 例22009年12月2日胸部X线片(两肺弥漫性毛玻璃样改变);图12 例22009年12月8日胸部X线片(两肺弥漫性毛玻璃样改变,较前无改善)
十二指肠溃疡伴食物嵌顿1例
(正文见第1735页

)
图1 胃镜下嵌顿食物团块的十二指肠球部
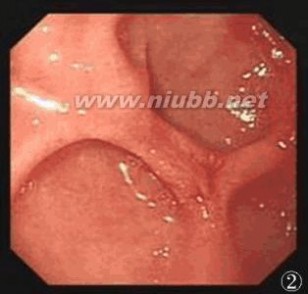
图2 胃镜下三枚较浅的假憩室
本文标题:色素失禁症-色素失禁症研究进展61阅读| 精彩专题| 最新文章| 热门文章| 苏ICP备13036349号-1